 VIII.
Psychoactive Drugs.
VIII.
Psychoactive Drugs.
VIII.
Psychoactive Drugs.
Outline
A. Sedatives and Hypnotics
1. Benzodiazepines
2. Barbiturates
B. Antipsychotic Drugs
1. Phenothiazines
2. Dibenzodiazepines
C. Anti-Depressants
1. Monoamine Oxidase Inhibitors
2. Serotonin
3. Tricyclic Anti-Depressants
4. Selective Serotonin Reuptake Inhibitors
5. Lithium
D. Stimulants
1. Amphetamine
2. Methylxanthines
A. Sedatives and Hypnotics
A sedative drug decreases activity, moderates excitement, and calms the recipient. A hypnotic drug produces drowsiness and facilitates the onset and maintenance of a state of sleep that resembles natural sleep in its electrocephalographic characteristics and from which the recipient may be easily aroused; the effect is sometimes called hypnosis. Sedation, pharmacological hypnosis, and general anesthesia are usually regarded as only increasing depths of a continuum of central nervous system (CNS) depression. Indeed, most sedative or hypnotic drugs, when used in high doses, can induce general anesthesia. One important exception is the benzodiazepines
The term benzodiazepine refers to the portion of the structure composed of a benzene ring (A) fused to a seven-membered diazepine ring (B). However, since all of the important benzodiazepines contain a aryl substituent (ring C) and a 1, 4-diazepine ring, the term has come to mean the aryl-1,4-benzodiazepines. There are several useful benzodiazepines available. The skeletal structure and two examples are shown below.
.
3-D
The effects of the benzodiazepines virtually all result from action of these drugs on the CNS, even when lethal doses are used. The most prominent of these effects are sedation, hypnosis, decreased anxiety, muscle relaxation, and anticonvulsant activity. As the dose of a benzodiazepine is increased, sedation progresses to hypnosis and hypnosis to stupor. They are used as sedatives, hypnotics, antianxiety agents (in panic disorder), anticonvulsants, muscle relaxants, in anesthesia and in alcoholism.
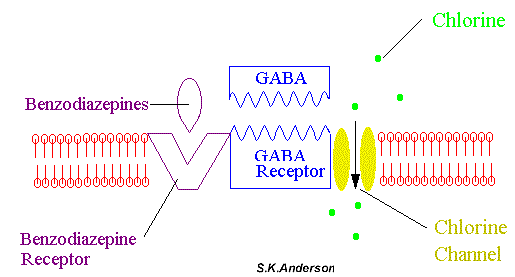
The actions of benzodiazepines are a result of potentiation of neural inhibition that is mediated by gamma-aminobutyric acid (GABA*). This view is supported by behavioral and electrophysiological evidence that the effects of benzodiazepines are reduced or prevented by prior treatment with antagonists of GABA or inhibitors of the synthesis of the transmitter. Benzodiazepine receptors are located on the alpha subunit of the GABA receptor (see figure below) located almost exclusively on postsynaptic nerve endings in the CNS (especially cerebral cortex). Benzodiazepines enhance the GABA transmitter in the opening of postsynaptic chloride channels which leads to hyperpolarization of cell membranes. That is, they "bend" the receptor slightly so that GABA molecules attach to and activate their receptors more effectively and more often.
The remarkable safety of the benodiazepines can be accounted for by the self-limited nature of neuronal depression that requires the release of an endogenous inhibitory neurotransmitter to be expressed. That is, they do not directly act to open chloride ion channels, and therefore, are not lethal in over dose as are barbiturates.
Benzodiazepines are highly lipid soluble. There is rapid diffusion into the CNS followed by redistribution to inactive tissue sites. Benzodiazepines have a high volume of distribution and rapidly cross the placenta. The duration of action is determined by rate of metabolism and elimination. Diazepam is not water soluble and is dissolved in propylene glycol; therefore it may cause venous irritation and thrombophlebitis. Diazepam also has unpredictable absorption after IM injection. Benzodiazepines are extensively bound to albumin.
General Info on Valium Valium Ads 1, 2, 3 General Info on Specific Benzodiazepines More General Info on Benzodiazepines
Information on Chlordiazepoxide
The barbiturates once enjoyed a long period of extensive use as sedative-hypnotic drugs; however, except for a few specialized uses, they have been largely replaced by the much safer benzodiazepines.
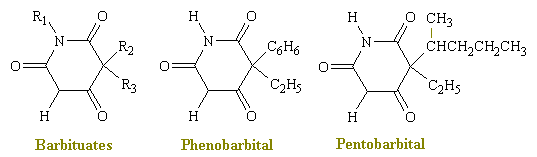
Barbiturates are CNS depressants and are similar, in many ways, to the depressant effects of alcohol. To date, there are about 2,500 derivatives of barbituric acid of which only 15 are used medically. The first barbiturate was synthesized from barbituric acid in 1864. The original use of barbiturates was to replace drugs such as opiates, bromides, and alcohol to induce sleep. Barbiturates are broken down chemically within the liver and eliminated via the kidneys at different rates according to their type. With regular use, the body develops a tolerance to barbiturates that translates into a need for larger and more frequent doses to attain the desired affect. However, while the tolerance increases in terms of realizing a desired effect, tolerance to the lethal level does not.
In general, structural changes that increase lipid solubility decrease duration of action, decrease latency to onset of activity, accelerate metabolic degradation, and increase hypnotic potency. Thus, large aliphatic groups at R2 confer greater activity than do methyl groups, but the compounds have a shorter duration of action; however, groups longer than seven carbons tend to have convulsive activity. Introduction of polar groups, such as ether, keto, hydroxyl, amino, or carboxyl groups, into alkyl side chains decreases lipid solubility and abolishes hypnotic activity.
Barbiturates facilitate GABA-ergic inhibition in a way that resembles some of the actions of the benzodiazepines, discussed above. However, barbiturates do not displace benzodiazepines from their binding sites. Instead, they enhance such binding by increasing the affinity for benzodiazepines; they also enhance the binding of GABA and its agonist analogs to specific sites in neural membranes. These effects are almost completely dependent upon the presence of chloride or other anions that are known to permeate through chloride channels and they are completely inhibited by picritoxin. (The picrotoxin group of toxins are naturally-occurring GABA antagonists which can cause death due to convulsions.)
While both barbiturates and benzodiazepines are capable of potentiating GABA-induced increases in chloride conductance, significant differences in their modes of action can be detected. Pentobarbital appears to increase the lifetime of the open state of the chloride channels that are regulated by GABA-ergic receptors; the magnitude of this effect more than offsets a barbiturate-induced decrease in the frequency of channel openings. By contrast, high concentrations of diazepam increase the frequency of channel openings with little effect on the lifetime of the open state. It is thought that barbiturates prolong the activation of the channel by decreasing the rate of dissociation of GABA from its receptor.
B. Antipsychotic Drugs
Schizophrenia comes in many varieties. One of the most common types is seen in the person who hears voices and has delusions of grandeur, intense fear, or other types of feelings that are unreal. Many schizophrenics are highly paranoid, with a sense of persecution from outside sources.
Schizophrenia appears to result from excessive excitement of a group of neurons that secrete dopamine in the behavioral centers of the brain, including in the frontal lobes. An alternative possibility is either hypersensitive or excess D2 dopamine receptors. Therefore, drugs used to treat this disorder decrease the level of dopamine excreted from these neurons or antagonize dopamine.
Dopamine has been implicated as a cause of schizophrenia because many patients with Parkinson's disease develop schizophrenic-like symptoms when they are treated with the drug L-DOPA. Also, drugs known to enhance central dopamine activity can worsen symptoms and even produce psychotic-like signs in normal individuals. It has been suggested that in schizophrenia, excess dopamine is secreted by a group of dopamine-secreting neurons whose cell bodies lie in the mesencephalon, medial to the substantia nigra. These neurons give rise to the so-called mesolinic dopaminergic system that projects nerve fibers to the medial and anterior portions of the limbic system, especially to the hippocampus, amygdala, anterior caudate nucleus, and portions of the prefrontal lobes. All of these are powerful behavioral control centers. An even more compelling reason for believing that schizophrenia is caused by excess production of dopamine is that many drugs that are effective in treating schizophrenia-such as chlorpromazine, haloperidol, and thiothizene-all decrease the secretion of dopamine at the dopaminergic nerve endings or decrease the effect of dopamine on subsequent neurons.
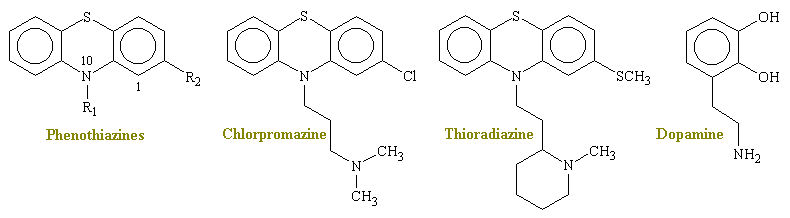
Doctors Schizophrenia Page Diagnosis Criteria Schizophrenia Home Page The phenothiazines as a class, and especially chlorpromazine, the prototype, are among the most widely used drugs in medical practice and are primarily employed in the management of patients with serious psychiatric illnesses. In addition, many members of the group have other clinically useful properties, including antiemetic, antinausea, and antihistaminic effects and the ability to potentiate analgesics, sedatives and general anesthetics.
Phenothiazine compounds were synthesized in Europe in the late nineteenth century as part of the development of aniline dyes such as methylene blue. In the late 1930s a derivative of phenothiazine was found to have antihistamine and a strong sedative effect and so the drug was introduced as into clinical anesthesia. It was noted that chlorpromazine by itself did not cause a loss of consciousness but produced only a tendency to sleep and a lack of interest in what was going on. These central actions became known as neuroleptic soon after.
Phenothiazine has a tricyclic structure in which two benzene rings are linked by a sulfur and a nitrogen atom (see figures below). Substitution of an electron-withdrawing group at R2 (but not at position 3 or 4) increases the efficacy of phenothiazines and other tricyclic congeners.
Neuroleptic drugs reduce initiative and interest in the environment, and they reduce displays of emotion or affect. Initially there may be some slowness in response to external stimuli and drowsiness. However subject are easily aroused, capable of giving appropriate answers to direct questions, and seem to have intact intellectual functions; there is no ataxia, incoordination, or dysathria at ordinary doses. Psychotic patients become less agitated and restless, and withdrawn or autistic patients sometimes become more responsive and communicative. Aggressive and impulsive behavior diminishes. Gradually (over a period of days). psychotic symptoms of hallucinations, delusions, and disorganized or incoherent thinking tend to disappear.
The most prominent observable effects of typical neuroleptic agents are strikingly similar. In low doses, operant behavior is reduced but spinal reflexes are unchanged. Exploratory behavior is diminished, and responses to a a variety of stimuli are fewer, slower, and smaller, although the ability to discriminate stimuli is retained. Conditioned avoidance behaviors are selectively inhibited, while unconditioned excape or avoidance responses are not.
Behavioral activation, stimulated environmentally of pharmacologically, is blocked. Feeding is inhibited. Most neuroleptics block the emesis and aggression induced by apomorphine--a dopaminergic agonist. In high doses, most neuroleptic agents induce characteristic cataleptic immobility that allows the animal to be placed in abnormal postures that persist. Muscle tone is altered, and ptosis (drooping of the eyelids) is typical. Even very high doses of most neuroleptics do not induce comma, and the lethal dose is extraordinarily high.
Mechanism. It is well established that benzodiazepines block dopaminergic receptors in the brain. There are three major central dopamine pathways: the nigrostriatal, which is affiliated with motor effects produced by antipsychotic drugs; the tuberoinfudibular, which is associated with the endocrine effects of neuroleptics; and the mesolimbic, which is the most likely to relate to the symptoms of schizophrenia. Of the three central dopamine receptor subtypes D1, D2 and D3, the D2 receptor, is believed to be most relevant to antipsychotic drug action. Most interesting, however, is that both D1 and D2 are altered in drug-naive schizophrenics. Though neuroleptic drugs are D1 and D2 antagonists, in vitro D2 effects are achieved at 103 lower concentrations.
D2 receptors are also located outside the blood brain barrier. One area is in the the chemoreceptor trigger zone of the medulla which is the reason that many of the phenothiazine drugs work as antiemetics and stop nausea.
Side Effects. There are several resulting syndromes which can occur from using antipsychotic drugs. A parkinsonian syndrome that may be indistinguishable from idiopathic parkinsonism my develop during administration of antipsychotic drugs. The most noticeable signs are rigidity and tremor at rest, especially involving the upper extremities. Tardive dyskenesia is a late-appearing neurological syndrome also associated with antipsychotic drug use. It is characterized by stereotypical involuntary movements consisting in sucking and smacking of the lips, lateral jaw movements, and fly-catching dartings of the tongue. There may be purposeless, quick movements of the extremities and slower, more dystonic movements and postures of the extremities, trunk, and neck may also be seen. All of these movements disappear during sleep as they do in parkinsonism. Symptoms of these conditions my persists indefinitely after discontinuation of the medication, although sometimes they disappear with time.
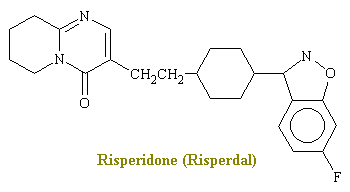
Dibenzodiazepine - Risperidone
In 1994 an addition tot he antipsychotic drugs is risperidone (Risperdal). This drug antagonises D2 and serotonin type 2 receptors. The drug also antagonizes for other receptors such as a adrenergic and histaminergic H1 receptors.
C. Anti Depressants
Major depression is the most common of the major mental illnesses, and it must be distinguished from normal grief, sadness, and disappointment. Major depression is characterized by feelings of intense sadness and despair, mental slowing and loss of concentration, pessimistic worry, agitation, and self-depreciation. Physical changes also occur, such as weight loss, decreased libido, and disruption of hormonal circadian rhythms.
Before the advent of psychotherapy in the 1950s, treatment of depression consisted of stimulants such as caffeine and amphetamines to ameliorate the depressive phases and barbiturates to allay agitation, anxiety, and insomnia. At best, such attempts at therapy may have offered transient relief to some patients. Suffering usually decreased little.
Neurobiology of Depression http://www.sciam.com/0189issue/0189wurtman.html
Monoamine Oxidase Inhibitors
Monoamine oxidase inhibitors were the first effective antidepressants used. These were discussed in detail in the section on Adrenergic Mechanisms and therefore will not be further discussed here.
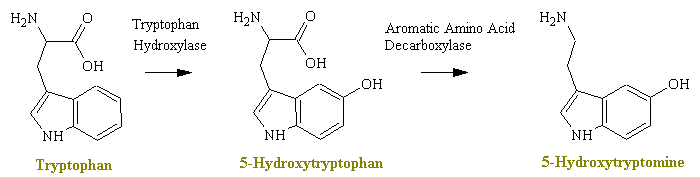
Serotonin (5-hydroxytryptamine or 5-HT) is a monoamine neurotransmitter found in cardiovascular tissue, in endothelial cells, in blood cells, and in the central nervous system. The role of serotonin in neurological function is diverse, and there is little doubt that serotonin is an important CNS neurotransmitter. The cell bodies for serotonergic neurons are found in the raphe region in the brainstem/pons region. Lesions of this area can be made using 5,6 or 5,7-dihydroxytryptamine in a similar manner to 6-hydroxydopamine and have helped define the CNS pathways for 5-HT.
The monoamine serotonin is itself a precursor for melatonin production in the pineal gland. The biosynthesis of serotonin from the amino acid tryptophan is similar to that found for the catecholamines, and 5-hydroxytryptophan can cross the BBB to increase central levels of 5-HT.
Although some of the serotonin is metabolized by monoamine oxidase to 5-hydroxyindole acetic acid, most of the serotonin released into the post-synaptic space is removed by the neuron through a reuptake mechanism inhibited by the tricyclic antidepressants and the newer, more selective antidepressants such as fluoxetine and sertraline.
Serotonin receptors are diverse and numerous. Over the past several years, over fourteen different serotonin receptors have been cloned and sequenced through molecular biological techniques. Overall, there are seven distinct families of 5-HT receptors, with as many as five within a particular family. Only one of the 5-HT receptors is a ligand-gated ion channel (the 5-HT3 receptor), and the other six families are all G protein-coupled receptors.
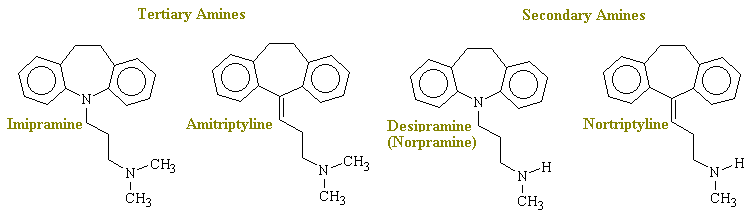
Tricyclic Anti-depressants
Imipramine, amitriptylin, and other closely related drugs are among the drugs currently most widely used for the treatment of major depression. Because of there structure ( see below). They are often referred to as the tricyclic antidepressants. Although these compounds seem to be similar to the phenothiazines chemically, the ethylene group of imiprimine's middle ring imparts dissimilar stereochemical properties and prevents conjegation of the rings, as occurs with the phenothiazines.
One might expect an effective antidepressant drug to have a stimulating or mood-elevating effect when given to a normal subject. Although this may occur with the MAOIs, it is not true of the tricyclic antidepressants. If a dose of imiprimine given to a normal subject, he feels sleepy and tends to be quieter, his blood pressure falls slightly, and he feels light headed. These drug effects are usually perceived to be unpleasant, and cause a feeling of unhappiness and increased anxiety. Repeated administration for several days may lead to accentuation of these symptoms and, in addition, to difficulty in concentrating and thinking. In contrast, if the drug is given over a period of time ( two to three weeks) to depressed patients an elevated mood occurs. For this reason, the tricyclics are not prescribed on an "as-needed" basis.
Mechanism. All tricyclic antidepressants in current use in the U.S. potentiate the actions of biogenic amines in the CNS by blocking its re-uptake at nerve terminals. However, the potency and selectivity for the inhibition of the uptake of norepinephrine, serotonin, and dopamine vary greatly among the agents. The tertiary amine tricyclics seem to inhibit the serotonin uptake pump, whereas the secondary amine ones seem better in switching off the NE pump (see table below). For instance, imipramine and amitriptyline are potent and selective blockers of serotonin transport with small effects on NE uptake, while desipramine and nortriptyline inhibit the uptake of norepinephrine and exert smaller effects on serotonin inhibition. None of these agents is very effective as an inhibitor of dopamine transport; this contrasts with the rather nonselective inhibitory actions of cocaine and amphetamine on the uptake of both norepinephrine and dopamine. These are poor antidepressants, despite the fact that it has a stimulant and even euphoriant effect in some people.
Drug Type of Amine Serotonin Norepinephrine Dopamine Amitriptyline tertiary ++++ + 0 Imiprimine tertiary +++ ++ 0 Doxepine tertiary ++ + 0 Desipramine secondary 0 ++++ 0 Maprotiline secondary 0 +++ 0 Nortriptyline secondary + ++ 0 Protriptyline secondary ? +++ 0 D-Amphetamine - 0 +++ ++++
More Info on Tricyclic Antidepressants
Selective Serotonin Reuptake Inhibitors
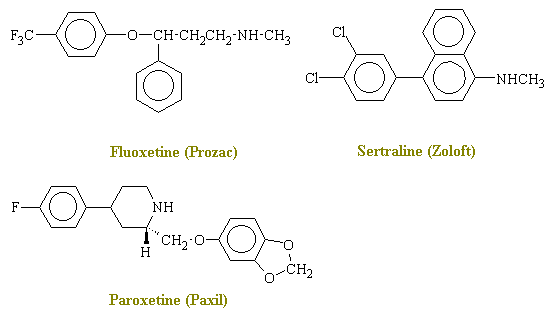
In recent years, selective serotonin reuptake inhibitors (SSRIs) have been introduced for the treatment of depression. Prozac is the most famous drug in this class. Lilly's sales of Prozac in 1993 exceeded 1 billion US dollars. Clomiprimine, fluoxetine (Prozac), sertraline and paroxetine selectively block the reuptake of serotonin, thereby increasing the levels of serotonin in the central nervous system. Note the similarities and differences between the tricyclic antidepressants and the selective serotonin reuptake inhibitors. The SSRIs generally have fewer anticholinergic side effects, but caution is still necessary when co-administering any drugs that affect serotonergic systems (e.g., monoamine oxidase inhibitors). Some of the newer, SSRIs (e.g., clomipramine) have been useful in the treatment of obsessive-compulsive disorders.
Here are some data to give you an idea of what transport systems are likely to be altered by the different antidepressants:
Drug Selectivity for 5-HT (Norepinephrine / Serotonin) clomiprimine 14 fluoxetine 54 fluvoxamine 160 paroxetine 280 sertraline 840 From: Hyttel J. Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). International Clinical Psychopharmacology. 9 Suppl 1: 19-26, 1994 Mar.
General Info on SSRIs
Lithium Salts
Lithium is widely used as Li2CO3 to control manic behavior in manic-depressive patients. No totally acceptable mechanism for its action exists. Postulations involve action that would likely adjust overactive catecholaminergic activity, which is the accepted occurrence in mania. Recent research has shown that Li+ is a glutamate reuptake inhibitor. Other research has shown that Li+ may inhibit glutamate stimulation of nerve cells.
Several studies showing that Li+ has some antidepressant effects are known. Some weak biphasic alterations of norepinephrine and serotonin turnover in the brain were established.
Amphetamines
Amphetamines were discussed under the topic of Adrenergic Mechanisms and therefore will not be further discussed here.
Methylxanthines
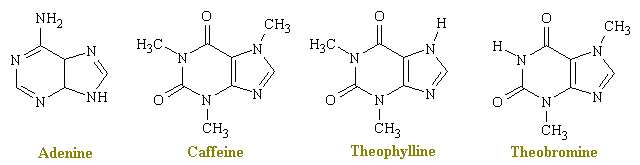
Caffeine, theophylline and theobromine share in common several pharmacological actions of therapeutic interest. They stimulate the central nervous system, act on the kidney to produce diuresis, stimulate cardiac muscle, and relax smooth muscle, notably bronchial muscle. Because the various xanthines differ markedly in the intensity of their action on various structures, one particular xanthine has been used more than another for a particular therapeutic effect. Since theobromine displays low potency in these pharmacological actions, it has all but disappeared from the therapeutic scene.
Caffeine, theophyline, and theobromine occur naturally in plants widely distributed geographically. Caffeine is found in the coffee bean, tea leaves, guarana, and other plants. It is probably the most-used of all psychoactive drugs. From the figure below, we can see that the methylxanthines have a structure which is very similar to adenine.
We have already discussed the role of the second messenger, cAMP, in the response to norepinephrine and epinephrine in the section on Adrenergic Mechanisms. The mechanism of action of caffeine and other methylxanthines is inhibiting the degradation of cAMP.
More Info on Theophylline Anything You Wanted to Know About Caffeine
Miscellaneous
questions
http://research.med.umkc.edu/pharm/tests/Psych.html
{kind=link}
{kind=link}
{kind=link}
{kind=link}