|
IV. Antimicrobial Drugs. |
Outline
A. Cell Wall Synthesis Inhibitors
1. b-lactam Antibiotics
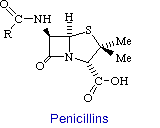
a. Penicillins
b. Cephalosporins
c. Mechanism
of Action
B. Protein Synthesis Inhibitors
1. Erythromycin
2. Tetracycline
C. Synthetic
Antibiotics
1. Sulfonamides
2. Antimalarial Drugs
D. Misuses of Antibiotics
The modern era of the chemotherapy of infection started with the clinical use of sulfanilamide in 1936. The "golden age" of antimicrobial therapy began with the production of penicillin in1941, when this compound was mass-produced and first made available for limited clinical trial. More than 30% of all hospitalized patients now receive one or more courses of therapy with antibiotics, and millions of potentially fatal infections have been cured. However, at the same time, these pharmaceutical agents have become among the most misused of those available to the practicing physician. One result of widespread use of antimicrobial agents has been the emergence of antibiotic-resistant pathogens, which in turn has created an ever-increasing need for new drugs. Many of these agents have also contributed significantly to the rising costs of medical care.
An antibiotic is any substance produced by a microorganism that is excreted to harm or kill another microorganism. Technically, antibiotics are microbial or fungal products. But these substances can be synthesized and mass produced in the laboratory to use against harmful microorganisms in the environment. Thus, the synthetic chemist has added greatly to our therapeutic armamentarium. Synthetic drugs such as isonaizid and theambutol represent important contributions for the treatment of tuberculosis. While many such antimicrobial agents are not properly termed antibiotics, since they are not produced by living organisms, little distinction should now be made between compounds of natural and synthetic origin.
Antibiotics can further be grouped under the broader heading of chemotherapuetic agents, chemical agents used to treat disease. Good chemotherapuetic agents are able to kill or inhibit the target pathogen without too much damage to the host organism. The basis for this selective toxicity lies in the differences between prokaryotic cells of microorganisms and our own eukaryotic cells. The prokaryotic cells of microorganisms differ in a number of ways from eukaryotic cells, such as absence of cell walls, different size of ribosomes, and details of metabolism. Thus, the goal of antibiotic therapy is to choose or design drugs that target these differences in host and pathogen cells.
A. Cell Wall Synthesis Inhibitors
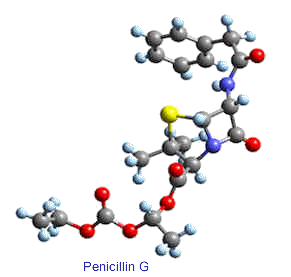
3-D structure of amoxicillin
Alexander Fleming loved to play, both in the laboratory and out. He always loved snooker and golf and had many whimsical variants on the rules. In the lab he made "germ paintings," in which he would draw with his culture loop using spores of highly pigmented bacteria, which were invisible when he made the painting, but when cultured developed into brightly colored scenes. He followed what Max Delbruck would later call the "principle of limited sloppiness." Fleming abhorred a tidy, meticulous lab; he left culture dishes lying around for weeks and would often discover interesting things in them. Though the story has been told in many sometimes conflicting ways, something like this resulted in the discovery of penicillin. He seems to have left a culture dish lying on the lab bench and then gone away on vacation. When he returned a few spores of an unusual mold had germinated on the plate. When he cultured the bacteria on the plate he found that they grew up to within a few centimeters of the mold, but there were killed. A crude extract of the mold was then shown to have antibacterial properties. Fleming made this discovery in 1928 and by 1929 had named it penicillin (he was told by a colleague that the mold was a type of Penicillium and "penicillozyme" must have seemed cumbersome).
Fleming continued to use penicillin in his lab but not with any great enthusiasm and certainly not to the exclusion of many other
projects. He never developed it into a clinically useful compound, though in 1929 he suggested that it might have important clinical applications. Because he was a bacteriologist and not a chemist, Fleming did not attempt to purify penicillin. He seems to have run into a dead end with penicillin and so during the 1930s, though he kept it in his lab, he did not do much with it. In the late 1930s Australian Howard Florey came to London to work with Charles Sherrington. He worked on lysozyme for a while and then became interested in penicillin. It was Florey, with Chain and other of his group that developed penicillin into a clinical antibiotic. They did this during 1940-41. Fleming, Florey, and Chain shared the 1945 Nobel Prize in Physiology for Medicine.
Fleming became world-famous for penicillin, and was rightly acknowledged as the father of modern antibiotics, but Florey was just as rightly miffed at being denied much of the credit for creating the powerful medical tool we now know. Evidence does not suggest that Fleming deliberately denied Florey his due credit, but Fleming's peculiar, dry sense of humor seems to have caused him not to deny even the wildest attributions to him.Multi-Media
movie of penicillin destroying bacteria illustration showing penicillin production polarized picture of penicillin under a microscope
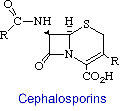
Cephalosporins are the second major group of b-lactam antibiotics. They differ from penicillins by having the b-lactam ring fused to a dihydrothiazine ring rather than a thiazolidine. The other difference, which is more significant from a medicinal chemistry stand point, is the existence of a functional group (R) at position 3 of the fused ring system. This now allows for molecular variations to be introduced at the 7-NH2 group, as in the penicillins, as well as to effect changes in properties by diversifying the moieties at position 3.
The first member of the newer series of b-lactams was isolated in 1956 from extracts of Cephalosporium acremonium, a sewer fungus. This species actually produced several antibiotics: cephalosporin C, cephalosporins P1 - P5 and penicillin N. This was also true with Fleming's Penicillium notatum. However, that was not discovered until research on the chemistry of penicillin was worked on. Cephalosporin has also been identified from other fungi such as Emericellopsis and Paecilomyces, two genera that are morphologically similar to Penicillium.
Like penicillin, cephalosporins are valuable because of their low toxicity and their broad spectrum of action against various diseases. In this way, cephalosporin is very similar to penicillin. Cephalosporins are one of the most widely used antibiotics, and economically speaking, has about 29% of the antibiotic market. The cephalosporins are possibly the single most important group of antibiotics today and are equal in importance to penicillin.
The structure and mode of action of the cephalosporins are similar to that of penicillin. They affect bacterial growth by inhibiting cell wall synthesis, in Gram-positive and -negative bacteria.
3-D structure of cefoxitin
Penicillin consists of a thiolidine ring fused to a b-lactam ring, to which a variable R group is attached by a peptide bond. This structure can undergo a variety of rearrangements, which accounts for the instability first encountered by Flemming. In particular, the b-lactam ring is very labile.
In 1957, it was shown that bacteria ordinarily susceptible to penicillin could be grown in its presence if a hypertonic medium were used. The organisms obtained this way are devoid of a cell wall and consequently lyse when transferred to a normal medium. Hence it was inferred that penicillin interferes with the synthesis of the bacterial cell wall. The cell walls of bacteria are essential for their normal growth and development. Peptidoglycan is a heteropolymeric component of the cell wall that provides rigid mechanical stability by virtue of its highly cross-linked lattice work structure, which prevents bacteria from bursting from their high internal osmotic pressure. The peptidoglycan is composed of glycan chains, which are linear strands of alternating pyranoside residues of two amino sugars (N-acetylgucosamine and N-acetylmuramic acid), that are cross-linked by peptide chains. In gram-positive microorganisms, the cell wall is 50 to 100 molecules thick, but it is only 1 or 2 molecules thick in gram-negative bacteria.
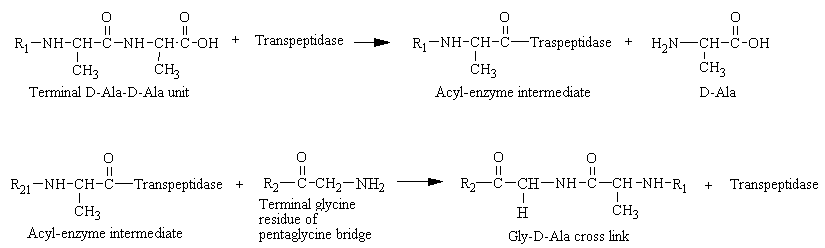
In 1965, it was deduced that penicillin blocks the last step in cell wall synthesis, namely the cross-linking of different peptioglycan strands. This cross-linking is accomplished by a transpeptidation reaction that occurs outside the cell membrane. The transpeptidase itself, called glycopeptide transpeptidase, is membrane bound. In the formation of the cell wall of Staphylococcus aureus, the transpeptidase normally forms an acyl-enzyme intermediate with the penultimate D-analine residue of the D-Ala-D-Ala-peptide. This covalent acyl-enzyme intermediate then reacts with the amino group of the terminal glycine in another peptide to form the cross-link (see figure below). Therefore, the end result is that the amino group at one end of a pentaglycine chain attacks the peptide bond between two D-analine residues in another peptide unit, a peptide bond is formed between glycine and one of the D-alanine residues, and the other D-alanine residues is released. It should be noted that bacteria cell walls are unique in containing D amino acids, which form cross-links by a different mechanism from that used to synthesize proteins.
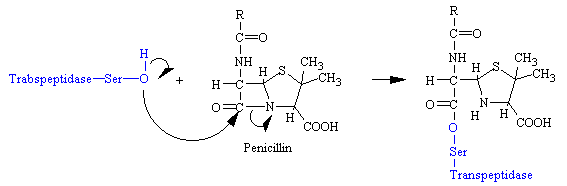
Penicillin mimics the D-Ala-D-Ala moiety of the normal substrate and is welcomed into the active site of the transpeptidase. Bound penicillin then forms a covalent bond with a serine residue at the active site of the enzyme. This penicilloyl-transpeptidase does not further react and the enzyme is irreversibly inhibited (see figure below).
The reason penicillin is so effective in inhibiting glycopeptide transferase is the four-membered b-lactam ring is strained, which makes it highly reactive. Also, the conformation of this part of penicillin is probably very similar to that of the transition state of the normal substrate, a species that interacts strongly with the enzyme.
Because of the high use of penicillin, some bacteria have developed resistance by producing molecules that can disable penicillin. Penicillinase is an enzyme produced by certain penicillin-resistant bacteria which reacts irreversibly with the b-lactam ring. Scientists have responded with other drugs that inturn react and disable penicillinase. One such drug is clavulanic acid. This compound irreversibly binds to penicillinase and prevent the enzyme from working. Therefore, sometimes clavulanic acid is given along with one of the semi-synthetic penicillins.
B. Protein Synthesis Inhibitors.
Erythromycin is an orally effective antibiotic discovered in 1952 in the metabolic products of a strain of Streptocyces erythreus, originally obtained from a soil sample collected in the Philippine Archepelago.
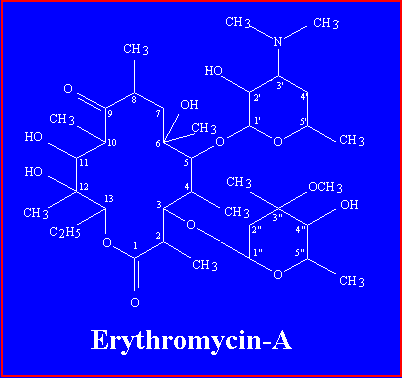
Erythromycin is one of the macrolide antibiotics, so named because they contain a many-membered lactone ring to which are attached one or more deoxy sugars. It is a white crystalline compound, soluble in water to the extent of 2 mg/ml. the structuras formula of erythromycine is a follows:
Erythromycin-A has a 14-membered macrolide ring, to which two sugars are attached: desosamine on carbon 5, and cladinose on carbon 3.
Crystal Image
Erythromycin may be either bacteriostatic or bactericidal, depending on the microorganism and the concentration of the drug. The bactericidal activity is greatest against a small number of rapidly dividing microorganisms and increases markedly as the pH of the medium is raised over the range of 5.5 to 8.5. the antibiotic is most effective in vitro against gram positive cocci such as Strep. pyogens and Strep. Pneumoniae. Resistant strains of these bacteria are rare and are usually isolated from populations of people who have been recently exposed to macrolide antibiotic. For example, in 1979, only 5% of group-A streptococcal strains isolated in Oklahoma were resistant to erythromycin, but 60% of such strains were found to be resistant in Japan. the difference likely reflects the wide use of erythromycin in Japan for respiratory infections.Mechanism of Action
Erythromycin and other macrolide antibiotics inhibit protein synthesis by binding to 50 S ribosomal subunits of sensitive microorganisms. (Humans do not have 50 S ribosomal subunits, but have ribosomes composed of 40 S and 60 S subunits). Certain resistant microorganisms with mutational changes in components of this subunit of the ribosome fail to bind the drug. The association between erythromycin and the ribosome is reversible and takes place only when the 50 S subunit is free from tRNA molecules bearing nascent peptide chains. The production of small peptides goes on normally in the presence of the antibiotic, but that of highly polymerized homopeptides is suppressed. Gram-positive bacteria accumulate about 100 times more erythromycin than do gram-negative microorganisms. The nonionized from of the drug is considerably more permeable to cells, and this probably explains the increased antimicrobial activity that is observed in alkaline pH.
Tetracycline
The development of the tetracycline antibiotics was the result of a systematic screening of soil specimens collected from many parts of the world for antibiotic-producing microorganisms. The first of these compounds, chlortetracycline, was introduced in 1948. Soon after there initial development, the tetracyclines were found to be highly effective against rickettsiae, a number of gram-positive and gram-negative bacteria, and the agents responsible for conjunctivitis, and psittacosis, and hence became known as "broad spectrum" antibiotics. They are also effective to agents that exert their effects on the bacterial cell wall, such as rickettsiae, Chlamydia, and amebae.
Chlortetracycline
In vitro, tetracycline drugs are primarily bacteriostatic and only multiplying microorganisms are affected. These drugs are closely related bacteriostatic antibiotics, similar in antibacterial spectrum and toxicity. The site of action of tetracyclines is the 30 S subunit of the ribosome, but at least two processes appear to be required for these antibiotics to gain access to the ribosomes of gram-negative bacteria. The first is passive diffusion through hydrophobic pores in the outer cell membrane. The second process involves an active transport system that pumps all tetracyclines through the inner cytoplasmic membrane. Although permeation of these drugs into gram-positive bacteria is less will understood, it too requires an active transport system. Once the tetracyclines gain access to the bacteria cell, they inhibit protein synthesis and bind specifically to 30 S ribosomes. They appear to prevent access of aminoacyl tRNA to the acceptor site on the mRNA-ribosome complex. This prevents the addition of amino acids to the growing peptide chain. Only a small portion of the drug is irreversibly bound, and the inhibitory effects of the tetracyclines can be reversed by washing. Therefore, it is probable that the reversibly bound antibiotic is responsible for the antibacterial action. These compounds also impair protein synthesis in mammalian cells at high concentrations; however the host cells lack the active transport system found in bacteria.
Tetracycline-resistant strains of pneumococci account for about 5% of isolates from pneumococcal pneumonia patients. Infections due to Group A beta-hemolytic streptococci should not be treated with a tetracycline, since as many as 25% of the organisms may be resistant when tested in vitro. Serious staphylococcal disease is also not a primary indication for tetracyclines. Bacterial resistance to one tetracycline is generally accompanied by cross-resistance to the others.
Pharmacology
The tetracyclines are variably absorbed after oral administration. Food interferes with absorption of tetracyclines, with the exception of doxycycline and minocycline. Absorption of tetracyclines is decreased in the presence of antacids containing aluminum, Ca, and Mg, and in preparations containing iron. The half-lives in plasma are about 8 h for oxytetracycline and tetracycline; 13 h for demeclocycline and methacycline; and 16 to 20 h for doxycycline and minocycline.
Tetracyclines penetrate into most tissues and body fluids. However, CSF levels are not reliably therapeutic. Minocycline, because of its high lipid solubility, is the only tetracycline that penetrates into tears and saliva in levels high enough to eradicate the meningococcal carrier state. All tetracyclines, except doxycycline, are excreted primarily in the urine by glomerular filtration, and their blood levels increase in the presence of renal insufficiency. Doxycycline is excreted mainly in the feces. All tetracyclines are partially excreted in bile, resulting in high biliary levels. They are then partially reabsorbed.
Sulfonamides
The sulfonamides are synthetic bacteriostatic antimicrobial agents with a wide spectrum encompassing most gram-positive and many gram-negative organisms. These drugs were the first effective chemotherapeutic agents to be employed systematically for the prevention and cure of bacterial infections in man. The considerable medical and public health importance of their discovery and their subsequent widespread use were quickly reflected in the sharp decline in morbidity and mortality figures for the treatable infectious decease. Before penicillin became generally available, the sulfonamides were the mainstay of antibacterial chemotherapy. While the advent of antibiotics has diminished the usefulness of the sulfonamides, they continue to occupy an important, although relatively small, place in the therapeutic armamentarium of the physician.
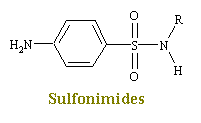
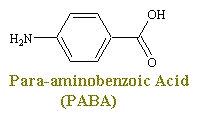
Sulfonimides are structural analogs and competitive antagonists of para-aminobenzoic acid (PABA), and thus prevent normal bacterial utilization of PABA for the synthesis of the vitamin folic acid. (The role of folic acid in RNA synthesis was already discussed in Anti Cancer Drugs). More specifically, sulfonamides are competitive inhibitors of the bacterial enzyme sulfihydropteroate synthase, which is responsible for the conversion of PABA into dihydrofolic acid, the immediate precursor of folic acid. Sensitive microorganisms are those that must synthesis their own folic acid; bacteria that can utilize preformed folic acid are not affected. Bacteriostasis induced by sulfonamides is counteracted by PABA competitively. Sulfonamides do not affect mamalian cells by this mechanism, since they require preformed folic acid and cannot synthesis it.
Pharmacology
Most sulfonamides are readily absorbed orally, the small intestine being the major site of absorption. Parenteral administration is difficult, since the soluble sulfonamide salts are highly alkaline and irritating to the tissues.
The sulfonamides are widely distributed throughout all tissues. High levels are achieved in pleural, peritoneal, synovial, and ocular fluids. CSF levels are effective in meningeal infections, but sulfonamides are rarely used for this indication. When given in pregnancy, high levels are achieved in the fetus. Sulfonamides are loosely and reversibly bound in varying degrees to serum albumin. Since the bound sulfonamide is inactive and nondiffusible, the degree of binding can affect antibacterial effectiveness, distribution, and excretion. The antibacterial action is inhibited by pus.
The sulfonamides are metabolized mainly by the liver to acetylated forms and glucuronides, both therapeutically inactive. Excretion is primarily renal by glomerular filtration with minimal tubular secretion or reabsorption.
The relative insolubility of most sulfonamides, especially their acetylated metabolites, may cause precipitation in the renal tubules. The more soluble analogs, such as sulfisoxazole and sulfamethoxazole, should be chosen for systemic therapy, and the patient must be well hydrated. To avoid crystalluria and renal damage, fluid intake should be sufficient to produce a urinary output of 1200 to 1500 mL/day. Sulfonamides should not be used in
renal insufficiency.
Antimalarial Drugs - Quinolines
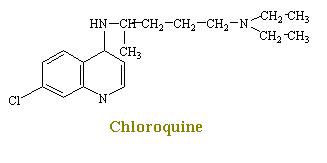
Chloroquine is one of the cheif agents for the chemotherapy of malaria. Although chlorouqine causes a number of effects that singly or in combination may relate to its promary mechanism of plasmodicidal action, this process is not yet shown. From early work, it has been found that chloroquine inhibits the incorporation of phosphate into RNA and DNA. Later it was shown that chloroquine combines strongly with double-stranded DNA. The drug is also reported to inhibit DNA polymerase activity markedly by combing with the DNA primer.
Basic Information Uses for Chloroquine Chloroquine Under Polarized Light
A common misuse of these agents is in infections that have been proven to be untreatable. The vast majority of the diseases due to the true viruses will not respond to any of the presently available anti-infective compounds. Thus, the antimicrobial therapy of measles, chickenpox, mumps, and at least 90% of infections of the upper respiratory tract is totally ineffective and, therefore, worse than useless
Fever of undetermined etiology may be of two types: one that is present for only a few days to a week and another that persists for an extended period. Both of these are frequently treated with antimicrobial agents. Most instances of pyrexia of short duration, in the absence of localizing signs, are probably associated with undefined viral infections, often of the upper respiratory tract, and do not respond to antibiotics. In the bulk of these cases, defervescence takes place spontaneously within a week or less. Studies of prolonged fever have shown that three common infectious causes are tuberculosis, often of the disseminated variety, hidden pyogenic intraabdominal abscess and infectious endocarditis. Also, the so-called collagen disorders and various neoplasms, especially lymphoma are frequently responsible for prolonged and significant degrees of fever. Various types of cancer, metabolic disorders, hepatitis, asymptomatic regional enteritis, atypical rheumatoid arthritis, and a number of other noninfectious disorders may present themselves as cases of fever of unknown etiology. The most rational approach to the problem of fever of unknown origin is not one that concentrates of the elevated temperature alone but one that involves a thorough search for its cause. The patient should not be unnecessarily exposed to chemotherapy in the hope that, if an agent is not effective, another one or combination of drugs will be helpful.
Because of misuse of antibiotics many strains of bacteria have become resistant to the effects of these drugs. Here are some examples of resistance that has occurred in staphylococcus species:
1. In 1952, almost 100% of Staphylococcus infections were susceptible to penicillin. By 1982, fewer than 10% of staph cases could be cured with penicillin. The resistance initially was due to one type of resistance mechanism, and alternative drugs were available. For example, in the late 1960’s, physicians switched to methicillin for staph infections.
2. In the early 1980’s, Staphylococcus strains were found that were resistant to penicillins, methicillin, naficillin, and cephalosporins. The source of these bacteria was ahospital. That is, the toughest cases of bacterial infections will be treated in hospitals, so that is where these highly resistant bacteria are most likely to be found. Approximately 19,000 hospital patients die each year due to nosocomial (hospital-acquired) bacterial infections.
3. In 1992, roughly 15% of all Staphylococcus strains in the U.S. were methicillin-resistant. By 1993, only one surefire Staphylococcus killer remained: vancomycin. However, vancomycin-resistant strains of other bacteria have been found, and now Staphylococcus has even become resistant to this antibiotic.
{kind=link}