III. Analgesics and
Anti-inflammatory Agents.
Outline
A. Aspirin-like
Drugs
1. Aspirin
2. Acetaminophen
3. Gold
B. Opiates, Opiate Antagonists and Opiate
Receptors
1. Opiates
2. Mechanism of Action
3. Basic Effects of Morphine
4. Opiate Antagonists
The anti-inflammatory, analgesic, and antipyretic drugs are a heterogeneous group of compounds, often chemically unrelated (although most of them are organic acids), which nevertheless share certain therapeutic actions and side effects. The prototype is aspirin; hence these compounds are often referred to as aspirin-like drugs.
All aspirin-like drugs are antipyretic, analgesic, and anti-inflammatory, but there are important differences in their activities. For example, acetaminophen is antipyretic and analgesic but is only weakly anti-inflammatory. The reason for the differences are not clear; variations in the sensitivity of enzymes in the target tissues may be important.
When employed as analgesics, these drugs are usually effective only against pain of low-to-moderate intensity, particularly that associated with inflammation. Aspirin drugs do not change the perception of sensory modalities other than pain. The type of pain is important; chronic postoperative pain or pain arising from inflammation is particularly well controlled by aspirin-like drugs, whereas pain arising from the hollow viscera is usually not relieved.
As antipyretics, aspirin-like drugs reduce the body temperature in feverish states. Although all such drugs are antipyretics and analgesics, some are not suitable for either routine or prolonged use because of toxicity; phenylbutaxone is an example.
This class of drugs finds its chief clinical application as anti inflammatory agents in the treatment of musculoskelatal disorders, such as rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis. In general, aspirin-like drugs provide only symptomatic relief from the pain and inflammation associated with the disease and do not arrest the progression of pathological injury.
Chemistry of Action
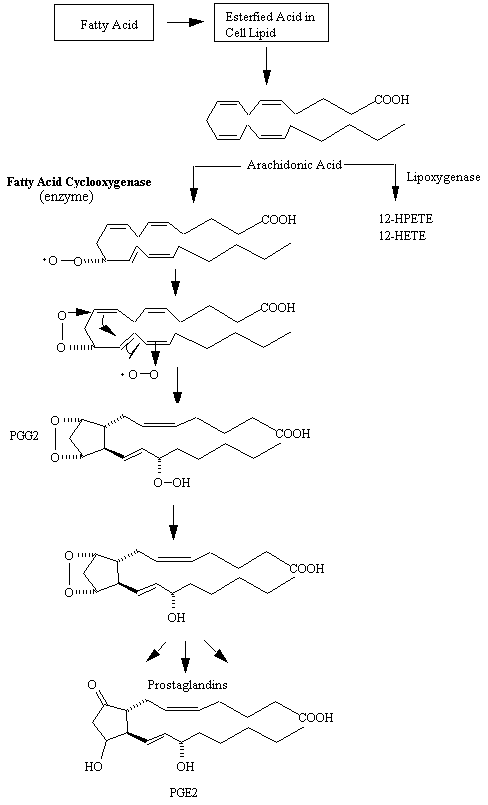
There has been substantial progress in elucidating the mechanism of action of aspirin-like drugs, and it is now possible to understand why such heterogeneous agents have the same basic therapeutic activities and often the same side effects. Indeed, their therapeutic activity appears to depend to a large extent upon the inhibition of a defined biochemical pathways responsible for the biosynthesis of prostaglandins (see figure below) and related autacoids.
Aspirin-like drugs inhibit the conversion of arachidonic acid to the unstable endoperoxide intermediate, PGG2, which is catalyzed by the cyclooxygenase. Individual agents have differing modes of inhibitory activity on the cyclooxygenase. Aspirin itself acetylates a serine at the active site of the enzyme. Platelets are especially susceptible to this action because (unlike most other cells) they are incapable of regenerating the enzyme, presumably because they have little or no capacity for protein biosynthesis. In practical terms this means that a single dose of aspirin will inhibit the platelet cyclooxygenase for the life of the platelet (8 to 10 days); in man a dose as small as 40 mg per day is sufficient to produce this effect. In contrast to aspirin, salicylic acid has no acetylating capacity and is almost inactive against cyclooxygenase in vitro. Nevertheless, it is as active as aspirin in reducing the synthesis of prostaglandins in vivo. The basis of this action and, thus, of the anti-inflammatory effect of salicylic acid is not clearly understood. Since aspirin is rapidly hydrolyzed to salicylic acid in vivo (half-life in human plasma, approximately 15 minutes), the acetylated and nonacetylated species probably act as pharmacologically distinct entities.
Most of the other common aspirin-like drugs are irreversible inhibitors of the cyclooxygenase, although there are some exceptions. For indomethacin, the mode of inhibition is particularly complex and probably involves a site on the enzyme different from that which is acetylated by aspirin.
Pain
Prostaglandins are associated particularly with the development of pain that accompanies injury or inflammation. Large doses of PGE2 or PGF2a, given to women by injection to induce abortion, cause intense local pain. Prostaglandins can also cause headache and vascular pain when infused intravenously in man. While the doses of prostaglandins required to elicit pain are high in comparison with the concentrations expected in vivo, induction of hyperalgesia occurs when minute amounts of PGE1 are given intradermally to man. Furthermore, in experiments in man where separate infusions of PGE1, bradykinin, or histamine caused no pain, marked pain was experienced when PGE1 was added to bradykinin or histamine. When PGE1 was infused with histamine, itching was also noted.
Fever
The hypothalamus regulates the set point at which body temperature is maintained. In fever, this set point is elevated, and aspirin-like drugs promote its return to normal. These drugs do not influence body temperature when it is elevated by such factors as exercise or increases in the surrounding temperature.
Fever may be a result of infection, tissue damage, inflammation, graft rejection, malignancy, or other disease states. A multitude of microorganisms can cause fever. There is evidence that bacterial endotoxins act by stimulating the biosynthesis and release by neutrophils and other cells of an endogenous pyrogen, a protein with a molecular weight of 10,000 to 20,000. The current view is that the endogenous pyrogen passes from the general circulation into the central nervous system, where it acts upon discrete sites within the brain, especially the preoptic hypothalamic area. There is evidence that the resultant elevation of body temperature is mediated by the release of prostaglandins and that aspirin-like drugs suppress the effects of endogenous pyrogen by inhibiting the synthesis of these substances. The evidence includes the ability of prostaglandins, especially PGE2, to produce fever when infused into the cerebral ventricles or when infected into the hypothalamus. Fever is a frequent side effect of prostaglandins when they are administered to a women as abortifacients. Moreover some studies have demonstrated an increase in prostaglandin-like substances in the cerebrospinal fluid when endogenous pyrogen is injected intravenously. The fever produced by the administration of pyrogen, but not that by prostaglandins, is reduced by aspirin-like drugs.
Side Effects
Aspirin is very useful, but it has many side effects and
therefore must be used carefully. Like most powerful drugs, an overdose of aspirin or
salicylates can be fatal. If a child or adult takes an overdose of aspirin, induce
vomiting to empty the unabsorbed medication from the stomach (if the person is still awake
and conscious). Obtain emergency medical care right away.
The most common side effects of aspirin are heartburn and other symptoms of stomach
irritation such as indigestion, pain, nausea, and vomiting. The stomach irritation may
lead to bleeding from the stomach, which may cause black stools. These symptoms may be
reduced by taking aspirin with meals, with an antacid, with a glass of milk, or by taking
enteric-coated or timed-release aspirin. Also, it is best not to take aspirin with alcohol
or coffee (or other beverages containing caffeine, such as tea or cocoa and many soft
drinks). Alcohol and caffeine make the stomach more sensitive to irritation. The non
aspirin salicylate preparations sometimes are less irritating to the stomach and may be
substituted for aspirin by your doctor.
A few people develop asthma, hay fever, nasal congestion, or hives from aspirin or
non-steroid anti-inflammatory drugs (NSAIDs). These people should never take aspirin, nor
should people who have active stomach or duodenal ulcers. Anyone who has ever had a peptic
ulcer should be very careful about taking aspirin because it can lead to a recurrence.
Aspirin is known to interfere with the action of the platelets. As a result, some people
who take a lot of aspirin experience easy bruising of the skin. Therefore, people who have
major bleeding problems should not take aspirin. Also, keep in mind that aspirin should
not be taken for 10-14 days before surgery (including surgery in the mouth) to avoid
excessive bleeding during or after the operation. These side effects probably depend on
aspirin-like drugs' ability to block endogenous prostaglandin biosynthesis.
Platelet function appears to be disturbed because aspirin-like drugs prevent the formation
by the platelets of thrombozane A2 (TXA2), a potent aggregating agent. This
accounts for the tendency of these drugs to increase the bleeding time.
Aspirin increases oxygen consumption by the body, increasing carbon dioxide production-an effect that stimulates respiration. Therefore, overdose with aspirin is often characterized by marked increases in respiratory rate, which cause the overdosed individual to appear to pant. This occurrence results in other, severe, metabolic consequences.
Prolongation of gestation by aspirin-like drugs has been
demonstrated in both experimental animals and the human female. Furthermore,
prostaglandins of the E and F series are potent uterotropic agents , and their
biosynthesis by the uterus increases dramatically in the hours before parturition.
It is thus hypothesized that prostaglandins play a major role in the initiation and
progression of labor and delivery.
High doses of salicylate may cause ringing in the ears and slight deafness.
Sometimes, however, these symptoms indicate mild overdose, which could become more
serious.
Aspirin and NSAIDs sometimes affect the normal function of the kidneys and aspirin-like
drugs promote the retention of salt and water by reducing the prostaglandin-induced
inhibition of both the reabsorption of chloride and the action of antidiuretic hormone.
This may cause edema in some patients with arthritis who are treated with an
aspirin-like drug.
Recent reports have said there could be a link between the use of aspirin and the
development of Reye's syndrome.
Reye's syndrome
is a rare but possibly fatal disease seen most often in children and teenagers. It usually
affects those recovering from chicken pox or a viral illness such as the flu. These
reports have raised concern in pediatricians (doctors who specialize in treating children)
and parents of children with arthritis who need to take large doses of aspirin to control
their disease.
In the U.S., about 10 to 20 thousand tons of aspirin are consumed each year; it is our most popular analgesic. Aspirin is one of the most effective analgesic, antipyretic, and anti-inflammatory agents.
| General Information | |
| More General Information | |
| Use as a Heart Medication |
Chemical structure
| GIF | |
| 3-D |
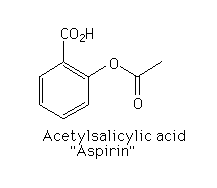
Acetaminophen is an effective alternative to aspirin as an analgesic and antipyretic agent. However, its anti-inflammatory effect is minor and not clinically useful. It is commonly felt that acetaminophen may have fewer side effects than aspirin, but it should be noted that an acute overdose may produce severe or even fatal liver damage. Acetaminophen does not inhibit platelet aggregation and therefore is not useful for preventing vascular clotting.
Side effects are usually fewer than those of aspirin; the drug produces less gastric distress and less ringing in the ears. However, as stated previously, overdose can lead to severe damage of the liver.
Acetaminophen has been proved to be a reasonable
substitute for aspirin when analgesic or antipyretic effectiveness is desired, especially
in patients who cannot tolerate aspirin. This might include patients with peptic
ulcer disease of gastric distress or those in whom the anticoagulant action of aspirin
might be undesirable.
Aspirin is often combined with acetaminophen in a single tablet for relief of arthritis
and other painful conditions. Sometimes other drugs such as caffeine, an antihistamine,
nasal drying agents, and sedatives are also added. Although some of these preparations may
have special uses for certain acute conditions such as a cold or a headache, they should
not be taken for a chronic (long-term) form of arthritis. If a combination is required,
each drug should be prescribed separately. The dose of each should be adjusted
individually to achieve the greatest benefit with the fewest side effects.
Researchers attribute the pain-relieving activity of acetaminophen to the drug's ability to elevate the pain threshold, although the precise mechanisms involved in this process have not been clearly identified. The antipyretic, or fever-reducing, effect of acetaminophen is far better understood. Research shows that the drug inhibits the action of fever-producing agents on the heat-regulating centers of the brain by blocking the formation and release of prostaglandins in the central nervous system. However, unlike aspirin and other NSAIDs, acetaminophen has no significant effect on the prostaglandins involved in other body processes.
| General Information | |
| More General Information |
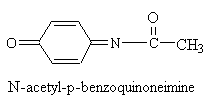
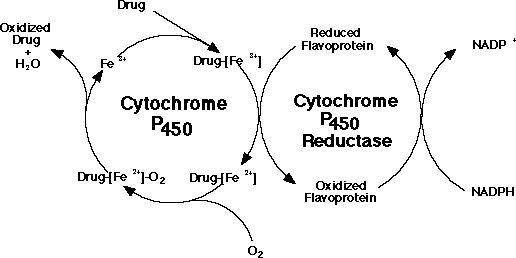
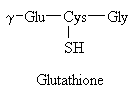
Despite claims to the contrary, stomach upset and hepatic toxicity are statistically as much a problem with acetaminophen as with aspirin-like drugs. Acetaminophen is normally metabolized in the liver and kidney by P450 enzymes. No toxicity is observed with therapeutic doses, however, after ingestion of large quantities (>2,000 mg/kg), a highly reactive metabolite, N-acetyl-p-benzoquinoneimine, is generated (see figure below). This species is electrophilic intermediate which is conjugated with glutathion to a non-toxic compound. Overdosing depletes glutithione and N-acetyl-p-benzoquinone reacts with nucleophilic portions (sulfhdryl groups) of critical liver cell protein. This results in cellular dysfunction and hepatic and renal toxicity. Antidote treatment consists of amino acid supplements to replenish glutathione. The P450 metabolizing enzymes differ somewhat in character between the liver and kidney. Factors that enhance renal toxicity include chronic liver disease, possibly gender, concurrent renal insults, and conditions that alter the activity of P450-metabolizing enzyme systems.
Other Aspirin-Like Drugs
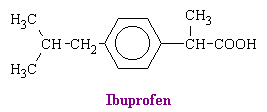
Other aspirin-like drugs include diflunisal, phenylbutazone, apazone, indomethacine, sulindac, fenamates, tolmetin, ibuprofen (see figure below), and piroxicam.
Gold is not of course an aspirin-like drug. However, its end effect is similar to aspirin, so it will be briefly considered here. Gold in elemental form has been employed for centuries as an antipruritic (anti itch medication) to relieve the itching palm. At present, gold treatment includes different forms of gold salts used to treat rheumatoid arthritis and related diseases. In some people, it helps relieve joint pain and stiffness, reduce swelling and bone damage, and reduce the chance of joint deformity and disability.
| general
information http://www.orthop.washington.edu/Bone%20and%20Joint%20Sources/xzzzzhzz1_2.html |
|
| more general information |
Chemistry
The significant preparations of gold are all compounds in which the gold is attached to sulfur. The three prominent drugs are aurothioglucose, auranofin, and gold sodium thiomalate.
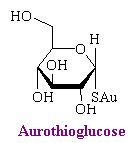
Side Effects
It takes months for gold compounds to leave the body. This
means that side effects to gold therapy may take some time to resolve. Sometimes side
effects even appear after the last gold injection.
Rash and a metallic taste in the mouth are side effects of gold injections that may not
seem serious at first. However, they are early warning signs for more serious reactions.
If either of these side effects develop, the health care provider should be contacted
promptly.
Some side effects may cause multiple symptoms, not all of which may occur.
Side effects with multiple symptoms are:
1. Low platelet count (thrombocytopenia). Most often this appears to be an
immunological disturbance that results in an accelerated degradation of platelets.
Symptoms may include: black, tarry stools, blood in urine, stool, or vomit, small red dots
on the skin, nose bleeds, unusual bruising or bleeding.
2. Anaphylaxis. Symptoms include sudden onset of the following soon after a gold injection: trouble swallowing, tightness in the throat, fainting, trouble breathing, wheezing, hives, swelling of the face, usually the lips or around the eyes, liver damage, abdominal pain for more than a few days, light colored stools, albuminuria, yellow eyes or skin, ulcerative colitis, severe abdominal pain or cramps, diarrhea that lasts more than a few days, blood in stool.
B. Opiates, Opiate Antagonists, and Opiate Receptors
The term opiate refers to any natural or synthetic drug that exerts actions upon the body similar to those induced by morphine, the major pain-relieving agent obtained from the opium poppy (Papaver somniferum). They were so highly regarded in the nineteenth century as remedies for pain, anxiety, cough, and diarrhea that some physicians referred to them as G.O.M.- `God's Own Medicine'. Opiates interact with what appear to be several closely related receptors, and they share some of the properties of certain naturally occurring peptides, the enkephalins, endorphins, and dynorphins.
The term opium
refers to the crude resinous extract obtained from the opium poppy. Crude opium
contains a wide variety of ingredients, including morphine and codeine, both of which are
widely used in medicine. The bulk of the ingredients of opium, however, consists of
such organic substances as resins, oils, sugars, and proteins that account for more than
75 % of the weight of the opium but exert little pharmacological activity. Morphine
is the major pain relieving drug found in opium, being approximately 10% of the crude
exudate. Codeine is structurally close to morphine (see figs below), although it is
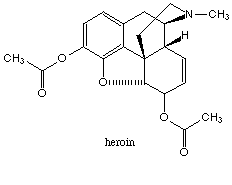
much less potent and amounts to only 0.5% of the opium extract. Heroin does not
occur naturally but is a semisynthetic derivative produced by a chemical modification of
morphine that increases the potency (see figs. below). It takes only 3 mg. of heroin
to produce the same analgesic effect as 10 mg of morphine. However, at these equally
effective doses, it may be difficult to distinguish between the effects of the two
compounds.
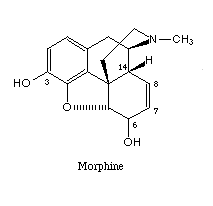
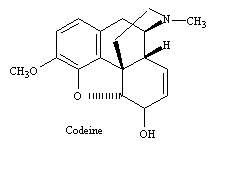

filled
structure of morphine
| Opiates | |
| More Info | |
| Endorphins | |
| Heroin and the Brain |
Studies of the binding of opioid drugs and peptides to specific sites in brain and other organs have suggested the existence of perhaps as many as eight types of receptors. In the CNS, there is reasonably firm evidence for four major categories of receptors, designated m, k, d, and s. To add confusion, there may well be subtypes of each of these receptors. Although there is considerable variation in binding characteristics and anatomical distribution among different species, inferences have been drawn from data that attempt to relate pharmacological effects to interactions with a particular constellation of receptors. For example, analgesia has been associated with both m and k receptors, while dysphoria or psychotomimetic (alteration of behavior or personality) effects have been ascribed to s receptors; based primarily on their localization in limbic regions of the brain, d receptors are thought to be involved in alterations of affective behavior. The actions of opioid drugs that are currently available have usually been interpreted with respect to the participation of only three types of receptors - m, k, and s; at each, a given agent may act as an agonist, a partial agonist, or an antagonist (see table). The m receptor is thought to meduate supraspinal analgesia, respiratory depression, euphoria, and physical dependance; the k receptor, spinal analgesia, miosis, and sedation; the s receptor, dysphoria, hallucinations, and respiratory and vasomotor stimulation.
It has been observed that opioids can selectively inhibit certain excitatory inputs to identified neurons. For example, the iontophoretic administration (the induction of an ionized substance through intact skin by the application of a direct current) of morphine into the substantia gelatinosa suppresses the discharge of spinal neurons in lamina IV of the dorsal horn that is evoked by noxious stimuli (e.g. heat) without changing responses to other inputs. While a postsynaptic action at discrete dedritic sites cannot be excluded, these findings suggest that opioids selectively inhibit the release of excitatory transmitters from terminals of nerves carrying pain related stimuli. In other situations, postsynaptic actions of opioids appear to be important. For example, application of opioids to neurons in the locus ceruleus reduces both spontaneous discharge and responses evoked by noxious stimuli. However, excitation of the neurons by antidromic stimulation (i.e. causing the neurons to fire backwards) is also suppressed, and the cells are hyperpolarized by the drugs.
Opioids have been observed to inhibit prostaglandin-induced increases in the accumulation of cyclic AMP in in brain tissue. Of potential relevance to mechanisms that underlie the phenomena of tolerance and withdrawal, the responses to prostaglandins recover in the continued presence of opioids.
Opioid-induced analgesia is due to actions of several sites within the CNS and involves several systems of neurotransmitters. Although opioids do not alter the threshold or responsivity of afferent nerve endings to noxious stimulation or impair the conduction of the nerve impulses along peripheral nerves, they may decrease conduction of impulses of primary afferent fibers when they enter the spinal cord and decrease activity in other sensory endings. There are opioid binding sites (m receptors) on the terminal axons of primary afferents within laminae I and II (substantia gelatinosa) of the spinal cord and in the spinal nucleus of the trigeminal nerve. Morphine-like drugs acting at this site are thought to decrease the release of neurotransmitters, such as substance P, that mediate transmission of pain impulses.
High doses of opioids can produce muscular rigidity in man, and both opioids and endogenous peptides cause catalepsy, circling, and stereotypical behavior in rats and other animals. These effects are probably related to actions at opioid receptors in the substania nigra and striatum, and involve interactions with both dopaminergic and GABA-ergic neurons.
The mechanism by which opioids produce euphoria, tranquility, and other alterations of mood remains unsettled. Microinjections of opioids into the ventral tegmentum activate dopaminergic neurons that project to the nucleus accumbens. Animals will work to receive such injections, and activation (or disinhibition) of these neurons has been postulated to be a critical element in the reinforcing effects of opioids and opioid-induced euphoria. However, the administration of dopaminergic antagonists does not consistently prevent these reinforcing effects. The neural systems that mediate opioid reinforcement in the ventral tegmentum appear to be distinct from those involved in the classical manifestations of physical dependence and analgesia.
CNS. Morphine exerts a narcotic action manifested by analgesia, drowsiness, changes in mood, and mental clouding. The major medical action of morphine sought in the CNS is analgesia, which may usually be induced by doses below those that cause other effects on the CNS, such as sedation or respiratory depression. The relief of pain by morphine-like opioids is relatively selective, in that other sensory modalities (touch, vibration, vision, hearing, etc.) are not inhibited. Patients frequently report that the pain is still present but that they feel more comfortable. Continuous dull pain is relieved more effectively than sharp intermittent pain, but with sufficient amounts of morphine it is possible to relieve even the severe pain associated with renal or biliary colic. In fact, its analgesic action appears to result not from a decrease of pain impulses into the CNS but from an altered perception of the painful stimuli.
Respiration. A second major action of morphine-like drugs is to depress respiration through interaction with m receptors located in the brainstem. At high doses, respiration may become so slow and irregular that life is threatened. In man, death from morphine poisoning is nearly always due to respiratory arrest. The primary mechanism of respiratory depression by morphine involves a reduction in the responsiveness of the brain stem respiratory centers to increases in carbon dioxide tension (PCO2). High concentrations of opioid receptors, as well, as endogenous peptides, are found in the medullary areas believed to be important in ventilatory control. Respiratory depression is mediated by a subpopulation of m receptors (m1), distinct from those that are involved in the production of analgesia (m2). Thus, a 'pure' m1-opioid agonist could theoretically produce analgesia with little respiratory depression.
Cough. Opiates suppress the "cough center" which is also located in the brainstem, the medulla. Such an action is thought to underlie the use of opiate narcotics as cough suppressants. Codeine appears to be particularly effective in this action and is widely used for this purpose.
Gastrointestinal Tract. The opiates have been used for centuries for the relief of diarrhea and for the treatment of dysentery, and these uses were developed long before these agents were used as analgesics or euphoiants. Opiates appear to exert their effect on the gastrointestinal tract primarily in the intestine, where peristaltic movements, which normally propel food down the intestine, are markedly diminished. Also, the tone of the intestine is greatly increased to the point where almost complete spastic paralysis of movement occurs. This combination of decreased propulsion and increased tone leads to a marked decrease in the movement of food through the intestine. This stasis is followed by a dehydration of the feces, which hardens the stool and further retards the advance of material. All these effects contribute to the constipating properties of opiates. Indeed, nothing more effective has yet been developed for treating sever diarrhea.
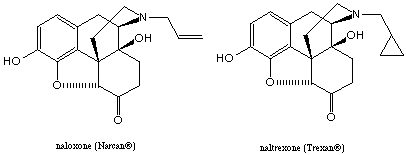
Naloxone, when administered to normal individuals, produces no analgesia, euphoria, or respiratory depression. However, it rapidly precipitates withdrawal in narcotic-dependent individuals. Naloxone antagonizes the actions of morphine at all its receptors; however its affinity for m receptors is generally more than ten fold higher than for k or d receptors.
The uses of naloxone include the reversal of the respiratory depression that follows acute narcotic intoxication and the reversal of narcotic-induced respiratory depression in newborns of mothers who have received narcotics. The use of naloxone is limited by a short duration of action and the necessity of parenteral route of administration.
Naltrexone became clinically available in 1985 as a new narcotic antagonist. Its actions resemble those of naloxone, but naltrexone is well is well absorbed orally and is long acting, necessitating only a dose of 50 to 100 mg. Therefore, it is useful in narcotic treatment programs where it is desired to maintain an individual on chronic therapy with a narcotic antagonist. In individuals taking naltrexone, subsequent injection of an opiate will produce little or no effect. naltrexone appears to be particularly effective for the treatment of narcotic dependence in addicts who have more to gain by being drug-free rather than drug dependant.
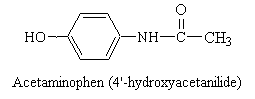
{kind=link}
{kind=link}
{kind=link}