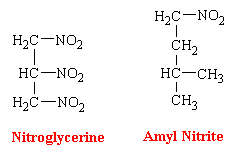VII. Cardiovascular Drugs
![]()
Outline
A. Antihypertensive Agents
1. Inhibition of Smpathetic Function
a. b-blockers
b. a-blockers
c. Reserpine
2. Diuretics
a. Carbonic
Anhydrase Inhibitors
b. Benzothiadiazines
B. Antianginal Drugs - Coronary Vasodilators
1. Organic Nitrates
a. Nitroglycerine
2. Viagra
C. Digitalis
D. Anticoagulants
1. Dicoumerol
Cardiovascular disease constitutes the largest single cause of death in the industrialized countries. As with cancer, which is a distant second in terms of mortality, cardiovascular disease morbidity increases with age, accounting for about two-thirds of all deaths in persons over 75.
Even though some diseases affect primarily the heart and other diseases effect the vascular system, they cannot be divorced from each other. This obvious interdependence makes a unified imperative. One of the major diseases, atherosclerosis, affects and ultimately damages the heart, kidneys, and other organs.
A. Antihypertensive Agents
Hypertension is generally defined as an elevation of systolic and/or arterial blood pressure and a value of 140/90 torr is generally accepted as the upper limit of normotension. Certain risk factors (e.g., hypercholesterolemia, diabetes, smoking, and a family history of vascular disease) in conjunction with hypertension predispose to arteriosclerosis and consequent cardiovascular morbidity. Patient populations with sustained diastolic blood pressures in the range of 105 to 129 torr are unequivocally benefited by effective reduction of blood pressure. The benefits of antihypertensive treatment are the avoidance of accelerated or malignant hypertension, a lower incidence of hypertensive renal failure, and a decrease in the incidence of hemorrhagic stroke and cardiac failure. Only recently has it been demonstrated that aggressive care of patients with mild diastolic hypertension can apparently reduce the incidence of myocardial infarction.
Inhibition of Sympathetic Function
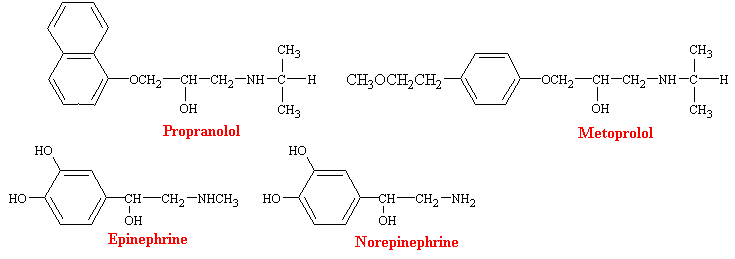
Since they where introduced in the 1960s, b-adrenergic blocking agents have become the most commonly used drugs for cardiovascular diseases. Propranolol was the first b-blocker to come into wide clinical use, and it remains the most important of these compounds. It is a highly potent, nonselective adrenergic blocking agent with not intrinsic sympathomimetic activity. However, because of its ability to block b receptors in bronchial smooth muscle and skeletal muscle, propranolol interferes with bronchodiation produced by epinephrine and with glycogenolysis, which ordinarily occurs during hypoglycemia. Thus, the drug is usually not used in individuals with asthma and must be used cautiously in diabetics who are receiving insulin. As a consequence, there has been a search for b-blockers that are cardioselective and a number of drugs now have been developed that exhibit some degree of specificity for b1-adrenergic receptors. Metaprolol is a prototype for these more specific drugs.
Much of the pharmacology of b-blockers can be deduced from a knowledge of the functions subserved by the involved receptors and the physiological conditions under which they are activated. Thus, b-receptor blockade has little effect on the normal heart with the subject at complete rest, but may have profound effects when sympathetic control of the heart is high, as during exercise. b-blockers decrease heart rate and cardiac output, prolongs mechanical systole, and slightly decreases blood pressure in resting subjects. Peripheral resistance is increased as a result of compensatory sympathetic reflexes, and blood flow to all tissues except the brain is reduced. Some of these drugs also have direct actions on cell membranes, which are commonly described as membrane stabilizing and local anesthetic. The local anesthetic potency of propranolol is about equal to that of lidocaine.
b-blockers are effective antihypertensive agents. Chronic treatment of hypertensive patients with b-adrenergic blocking agents results in a slowly developing reduction in blood pressure. Several mechanisms have been proposed for the efficacy of b-blockers in the management of hypertension. Reduction in cardiac output occurs rather promptly after administration of b-blockers. However, the hypotensive effects of b-blockers does not appear as rapidly. The release of renin from the juxtaglumarular apparatus is stimulated by adrenergic agonists, and this effect is blocked by drugs such as propranolol. Also, b-adrenergic agonists are known to increase modestly the release of norepinephrine from adrenergic nerve terminals, which causes vasoconstriction. b-blockers block this effect, and such impairment of the release of norepinephrine following sympathetic nerve stimulation might contribute to the antihypertensive effects of the drugs.
3-D Structure of Propranolol More Info on b1 and b2 Receptors
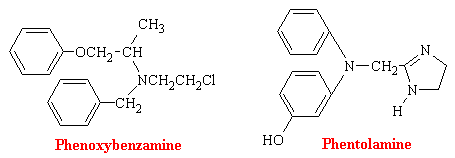
a-Adrenergic blocking agents exist in most blood vessels, particularly in coetaneous resistance vessels. Since their stimulation leads to constriction and therefore blood pressure elevation, it stands to reason that blocking such stimulation would lead to a diminution of blood pressure. Phenoxybenzamine binds covalently to the a receptor and produces an irreversible type of blockade. Phentolamine and tolazoline bind reversibly and antagonize the actions of sympathomimetic amines competitively.
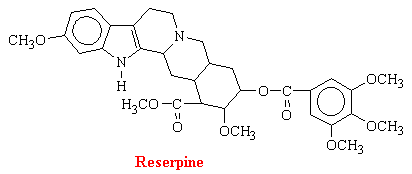
Rauwofia serpentina is a climbing shrub indigenous of India and neighboring countries. In 1954, it was reported that rauwolfia or reserpine was helpful in the treatment of psychotic patients because it acts centrally to produce characteristic sedation and a state of indifference to environmental stimuli; effects which resemble phenothiazines (discussed in Psychoactive Drugs). Subsequent discovery of the ability of rauwolfia alkaloids and related compounds to deplete biogenic amines from storage sites in the body initiated a great number of investigations directed at elucidating the interaction between these amines and reserpine. There are a number of rauwolfia alkaloids with complex structures. The structure of reserpine is as follows:
It is clear that reserpine interferes with intracellular storage of catecholamines, but the amounts of reserpine in tissues are much too small to assume a stoichiometric displacement. Reserpine antagonizes the uptake of norepinephrine by isolated chromaffin granules by inhibiting the ATP-Mg2+-dependent uptake mechanism of the granule membrane. The drug also binds to the vesicular membranes for days, accounting for the irreversibility of the process.
Reserpine depletes stores of catecholamines in many organs, including the brain and adrenal medulla, and most of its pharmacological effects have been attributed to this action. Since it is the reuptake and not the release of catecholamines that is inhibited, the existing pool must be fully depleted before antihypertensive effects become apparent. Also,, after administration of relatively large doses, reserpine causes a transient sympathomimetic effect followed by a slowly developing fall in blood pressure frequently associated with bradycardia. For normal doses, reduced concentrations of catecholamines can be measured within a hour after administration of reserpine, and depletion is maximal at 24 hours. Most of the catecholamine is deaminated intraneuronally, and pharmacological effects of the released mediator are minimal unless MAO has been inhibited.
The decrease in norepinephrine synthesis induced by reserpine is the block of dopamine uptake into storage granules that contain the enzyme dopamine b-hydroxylase (see norepinephrine synthesis). Furthermore, the increased concentration of free catecholamine presumably feeds back to inhibit tyrosine hydroxylase, since norepinephrine competes with the pterin cofactor for the enzyme.
Supersensitivity to catecholamines is observed following chronic administration of reserpine. The site of change is presumably postjunctional and may by due to alterations of the adrenergic receptors. Such adaptive change is usual following chronic deprivation of transmitter.
Reserpine is used as an effective antihypertensive agent, particularly when used with other agents such as diuretics. Its low cost, once-daily administration, and minimal change in effect when compliance is erratic make it useful as an agent for long-term treatment of patients with uncomplicated mild hypertension. However, reserpine causes mental depression in 25% of patients.
A diuretic is a substance that increases the rate of urine volume output, as the name implies. Most diuretics also increase urinary excretion of solutes, especially sodium and chloride. In fact, most diuretics that are used clinically act by decreasing the rate of sodium reabsorbtion from the nephron tubules, which in turn causes naturesis and this in turn causes diuresis. The most common clinical use of diuretics is to reduce extracellular fluid volume, especially in diseases associated with edema and hypertension.
Carbonic Anhydrase Inhibitors
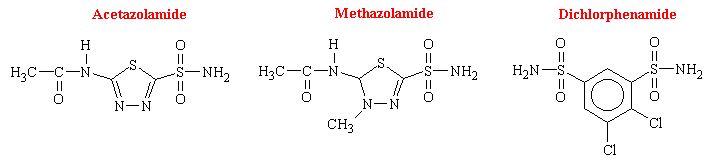
Soon after the introduction of the sulfonamides as antibacterial agents in the 1930s, changes in the electrolyte balance of patients were noted as was systemic acidosis accompanied by an alkalization of the urine due to increased rate of HCO3- excretion. Proposals by several researchers established that inhibition of the enzyme carbonic anhydrase (CA) accounted for the electrolytic imbalances produced. Since the antibacterial sulfonamides were relatively weak inhibitors, a successful search for more potent CA inhibition ensued. Acetazolamide became the first successful drug introduced into clinical use.
Among the enormous number of sulfonamides that have been synthesized and tested, acetazolamide has been studied the most extensively as an inhibitor of carbonic anhydrase. The other drugs of this class that are available in the U.S. are dichlorphenamide and methazolamide. There structural formulas are as follows:
Mechanism of Action. The kidneys control acid-base balance of the body by excreting either an acidic or a basic urine. The overall mechanism by which the kidneys accomplish this is as follows: Large numbers of bicarbonate ions are filtered continuously into the nephron tubules of the kidneys, and if they are excreted into the urine, this removes base from the blood. On the other hand, large numbers of hydrogen ions are also secreted into the tubular lumen by the tubular epithelial cells, thus removing acid from the blood.
Bicarbonate ions enter the tubular lumen of the kidney nephron with the glumerular filtrate. Bicarbonate ions do not readily permeate the luminal membranes of the renal tubular cells; therefore, bicarbonate ions that are filtered by the glomerulus cannot be directly reabsorbed. Instead, bicarbonate is reabsorbed by a special process in which it first combines with hydrogen ions to from H2CO3, which eventually becomes CO2 and H2O (see figure below).
This reabsorbption of bicarbonate ions is initiated by a reaction in the tubules between bicarbonate ions filtered at the glomerulus and hydrogen ions secreted by the tubular cells. The H2CO3 formed then dissociates into CO2 and H2O. The CO2 can move easily across the tubular membrane; therefore, it instantly diffuses into the tubular cell, where it recombines with H2O, under the influence of carbonic anhydrase, to generate new a H2CO3 molecule. This H2CO3 in turn dissociates to form bicarbonate ion and hydrogen ion. The hydrogen ions are secreted from the cell into the tubular lumen, by sodium-hydrogen pump, in exchange for sodium. The bicarbonate ions together with the exchanged Na+, then enter the peritubular blood supply. The hydrogen ion, now in the tubular lumen, combines with another bicarbonate ion to form H2CO3, which then again dehydrates to CO2, which reenters the tubular cell. The net result is reabsorbtion of most of the bicarbonate. Some of these concepts can be seen here in an animated nephron (you must have the shockwave plugin to view this). animated
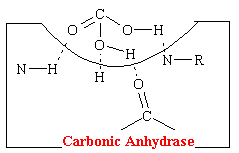
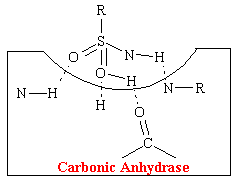
A hydrogen-bonding mechanism that acts competitively explains the action of acetazolamide-like carbonic anhydrase inhibitors that have diuretic properties. Carbonic acid is the normal substrate that fits into a cavity of and complexes with the enzyme carbonic anhydrase. This complex is strongly stabilized by four hydrogen bonds (see figure below).
The acetazolamide-like drugs fit into the cavity of the enzyme also effectively bond, presumably to the same four areas by hydrogen bonds (see figure below).
Thus these sulfonamide agents competitively prevent the carbonic acid from binding at this site which inhibits the reabsorption of NaHCO3 and H20. More than 99% of the enzyme activity in the kidney must be inhibited before physiological effects become apparent.
Following the administration of acetazolamide, the urine volume promptly increases. The urinary excretion of bicarbonate and fixed cation, mostly sodium (also potassium) and the normally acidic pH becomes alkaline. As a result, the concentration of bicarbonate in the extracellular fluid decreases and metabolic acidosis results. The urinary concentration of chloride falls.
The presence of carbonic anhydrase in a number of intraocular structure, including the cilliary processes, and the high concentration of bicarbonate in the aqueous humor have focused attention on the role that the enzyme might play in the secretion of aqueous humor. acetazolamide reduces the rate of aqueous humor formation; intaocular pressure in patients with glaucoma is correspondingly reduced.
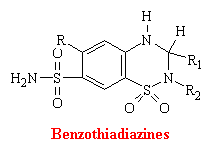
Thiazines and related compounds comprise the most frequently used antihypertensive agents in the United States. They were synthesized as an out growth of studies on carbonic anhydrase inhibitors. Thiazides act directly of the kidney to increase the excretion of sodium chloride and an accompanying volume of water. Also many of these drugs act as corbonic anhydrase inhibitors. At the molecular level, it is unknown how benzothiadiazines inhibit the reuptake of sodium chloride.
Chlorothiazide under the microscope.
B. Antianginal Drugs: Coronary Vasodilators
Angina pectoris, or ischemic heart disease, is the name given to the symptomatic oppressive pain resulting from myocardial ischemia. In simplest terms it results when the oxygen demand of myocardial tissues exceeds the circulatory supply. Once a local anemia due to an obstruction exists, biochemical changes are inevitable. Metabolic products will accumulate in the area, contractility declines, and NE release occurs from sympathetic neurons. The end result is pain. The underlying pathology of typical angina is usually advanced atherosclerosis and stenosis of the coronary vasculature which causes localized oxygen starvation. Episodes are be precipitated by emotional stress or exercise, but they usually cease rapidly with rest or nitroglycerin. In contrast, variant angina is caused by vasospasm of the coronary vessel and may or may not be associated with severe atherosclerosis. Patients with variant angina develop chest pain while at rest.
Until recently, many of the drugs used to prevent anginal attacks were no more effective than a placebo. In fact. the use of placebos has been reported to relieve symptoms in as many as 50% of patients with angina pectoris. For over a century, however, nitroglycerin has been known to be useful to prevent or relieve acute anginal attacks. More recently, the efficacy of b-adrenergic antagonists has been established for the long-term prophylaxis of typical angina. In addition, the calcium channel blockers appear to be effective for the treatment of vasospastic angina. While antianginal agents provide only symptomatic treatment, administration of these drugs do appear to decrease the incidence of sudden death associated with myocardial ischemia and infarction.
Organic Nitrates
The organic nitrates and nitrites are dilators of arterial and venous smooth muscle. The venodilation results in decreased left and right ventricular end-diastolic pressure, which are greater on a percentage basis than is the decrease in systemic arterial pressure. Net systemic vascular resistance is usually relatively unaffected; heart rate is unchanged or slightly increased; and pulmonary vascular resistance is consistently reduced. These drugs correct the inadequacy of myocardial oxygenation by increasing the supply of oxygen to ischemic myocardium by direct dilatation of the coronary vasculature and by decreasing the oxygen demand for oxygen by a reduction in cardiac work. The latter results from the decrease in vascular pressure enabling the heart to pump blood easier.
Glyceryl Trinitrate
Glyceryl trinitrate, or nitroglycerine, is a dense sweet-smelling oil that is highly explosive. The most utilized dosage form of nitroglycerin has been the sublingual tablet. Buccal absorption is rapid, offering almost instantaneous relief of sufficient duration (<30 min) for the emergency. Nevertheless, because of the valuable properties of nitroglycerin is now known to have in angina pectoralis, continuous blood levels of the drug are desirable. Therefore, different and innovative dosage forms are being developed. Because nitroglycerin is efficiently absorbed through the skin, this has led to the introduction of nitroglycerin skin patches. These patches contain the drug in a form which results in its continuous release.
Mechanism of Action
Nitric oxide (NO) has been shown to be an important messenger in many signal transduction processes. This free radical gas is naturally produced endogenously from arginine in a complete reaction that is catalyzed by nitric oxide synthetase (NOS).
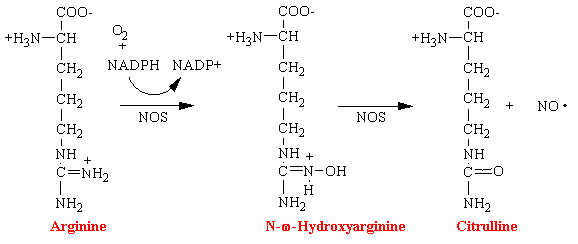
Nitroglycerin, nitrites, and organic nitrates also lead to the formation of the reactive free radical NO and is the basis of their mechanism of action. However, it is not known the exact enzyme that converts these drugs into NO.
Nitric oxide diffuses freely across membranes but has a short life, less than a few seconds, because it is highly reactive. Nitric oxide then activates a heme prosthetic group on the enzyme guanylate cyclase in the cell membrane. The heme molecule is, in effect, functioning as an extremely sensitive detector of NO. A portion of the enzyme protrudes to the interior of the cell and causes the formation of cyclic guanosine monophosphate (cGMP), a so-called second messenger. The cGMP in turn has many effects, one of which is to change the degree of phosphorylation of several enzymes that indirectly inhibit contraction. Especially, the pump that pumps calcium ions from the sarcoplasm into the sarcoplasmic reticulum in activated as well as the cell membrane pump that pumps calcium ions out of the cell itself; these effects reduce the intracellular calcium ion concentration, thereby inhibiting contraction.
General Info On Nitroglycerin History of Nitroglycerin Use Nitric Oxide Home Page Viagra, an experimental heart medication with a great side effect.
More on the Chemistry of Viagra Interactions of Viagra and Nitroglycerin
C. Digitalis
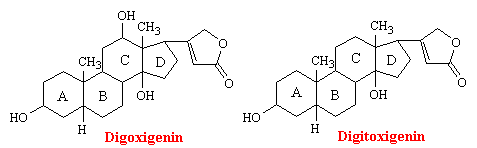
Digitalis is the dried leaf of the foxglove plant, Digitalis purpurea. Seeds and leaves of a number of other species also contain active cardiac principles. The two molecules in digitalis responsible for its pharmacological activity are digoxigenin and digitoxigenin. As you can see from the structures below, these genins are chemically related to sex and adrenocortical hormones.
Digitalis has a powerful action of the myocardium that is unrivaled in value for the treatment of heart failure. It is also used to slow the ventricular rate in the presence of atrial fibrillation and flutter. The main pharmacodynamic property of digitalis is its ability to increase the force of myocardial contraction. The beneficial effects of the drug in patients with heart failure - increased cardiac output; decreased heart size. venous pressure, and blood volume; diuresis and relief of edema - are all explained on the basis of increased contractile force, a positive inotropic action.
The most important component to the positive inotropic effect of digitalis direct inhibition of the membrane-bound Na+, K+-activated ATPase, which leads to an increase in intracellular [Ca+]. It seems clear that digitalis, in therapeutic concentrations, exerts no direct effect on the contractile proteins or on the interactions between them. Also, it seems most unlikely that the positive inotropic effect of digitalis is due to any action of the cellular mechanisms that provide the chemical energy for contraction. The hydrolysis of ATP by the Na+, K+-ATPase provides the energy for the so-called sodium-potassium pump - the system in the sarcolemma of cardiac fibers that actively extrudes sodium and transports potassium into the fibers. Digitalis drugs bind specifically to the Na+, K+-ATPase, inhibit its enzymatic activity, and impair the active transport of these two monovalent cations. As a result, there is a gradual increase in intracellular [Na+] and a gradual decrease in internal [K+]. These changes are small at therapeutic concentrations of the drug. It is judged to be crucially related to the positive inotropic effect of digitalis. This is so because in cardiac fibers, intracellular Ca2+ is exchanged for extracellular Na+ by a different transmembrane pump. When internal [Na+] is increased because of inhibition of the Na+, K+ pump by digitalis, the exchange of extracellular Na+ for intracellular Ca2+ is diminished, and internal [Ca2+] increases. The probable consequence of this is an increased store of Ca2+ in the sarcoplasmic reticulum and, with each action potential, a greater release of Ca2+ to activate the contractible apparatus.
Interactions of Na+, K+-ATPase with its various substrates are complex. Thus binding affinities with ATP, cofactor MG2+, Na+, K+, and a digitalis glycoside are all important to the overall effect. It is now accepted that the digitalis receptor is one or more of the conformations of Na+, K+-ATPase that occur during the ion pump's operation, possibly during a state in which the drug helps to stabilize one of the intermediate states of the enzyme.
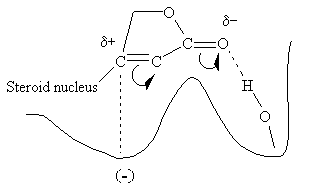
Evidence suggests that the entire glycoside molecule participates in the proposed drug-receptor interaction. The steric relationship of the lactone ring to the steroid nucleus is absolute. The double bond is also critical since saturation results in an almost total loss of activity. The required stereochemical positioning of rings C and D in relation to each other (cis) and of A and B (cis), and the configuration of the OH at C-14 (between rings C and D) have all been established. The figure below is a highly simplified version of a proposed interaction of the lactone side chain with such a digitalis receptor.
It is believed that the polarized carbonyl group, with its electron-rich oxygen, hydrogen bonds to a hydroxyl group on the enzyme's surface, probably to a serine residue, and the carbon atom bonded to the steroid nucleus is attracted to an electron dense site at a secondary location.
More Info on Digitalis
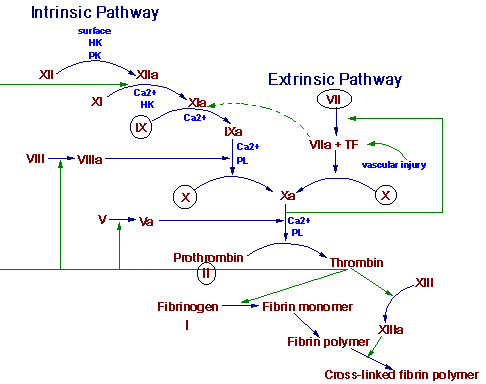
From any viewpoint blood is chemically the most complex tissue in the body. In addition to the multitude of cells and platelets, it contains inorganic ions, various plasma proteins, hormones, lipids, vitamins, a large variety of enzymes, nucleic acid breakdown products, a large number of unknown types of environmentally ingested chemical at varying stages of metabolic conversion, gases, and water. Among these is a group of more than a dozen chemical factors that will cause the blood to coagulate when properly triggered.
Hemostasis is the spontaneous arrest of bleeding from damaged vessels. Precapillary vessels contract immediately when cut. Within seconds, thrombocytes are bound to the exposed collagen of the injured vessel. Platelets also stick to each other and a viscous mass is formed. This platelet plug can stop bleeding quickly, but it must be reinforced by fibrin for long-term effectiveness. This reinforcement is initiated by the local stimulation of the coagulation process by the expose collagen of the cut vessel and the released contents and membranes of platelets. The two pathways of blood coagulation are shown in the figure below. The circled clotting factors are dependent on vitamin K for their activation.
Thrombogenesis is an altered state of hemostasis. An intravascular thrombus results from a pathological disturbance of hemostasis. The arterial thrombus is initiated by the adhesion and the release of circulating platelets to a vessel wall. This initial adhesion and the release of adenosine diphosphate (ADP) from platelets is followed by platelet-platelet aggregation. The thrombus grows to occlusive proportions in the areas of slower arterial blood flow. A platelet plug formed solely by platelet interaction is unstable. After the initial aggregation and viscous metamorphosis of platelets, fibrin becomes an important constituent of a thrombus. Production of thrombin occurs by activation of the reactions of blood coagulation at the site of the platelet mass. This thrombin stimulates further platelet aggregation not only by inducting the release of mote ADP from the platelets but also by stimulating the synthesis of prostaglandins, which are more powerful than ADP.
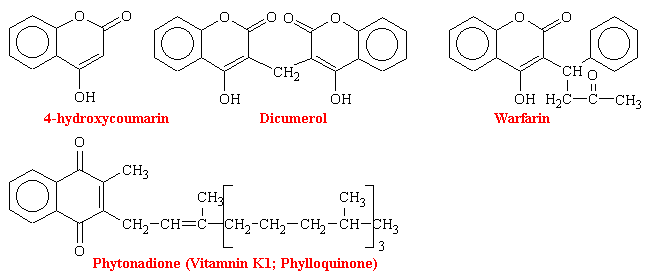
The first orally active anticoagulant, dicoumerol, which is a molecule consisting of two 4-hydroxycoumarin moieties bonded at their 3-position via a CH2 bridge, was isolated from decomposed yellow sweet clover. It was discovered and identified because of hemorrhagic death of cattle ingesting this improperly stored feed in the early 1920s (sweet clover disease). This followed demonstration that oral use of this compound increased clotting time and decreased the incidence of post surgical intravascular thrombus formation.
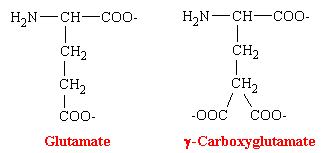
The oral anticoagulants are antagonists of vitamin K. Their administration to man or other animals leads to the appearance of precursors of the four vitamin K-dependent clotting factors in plasma and liver. These precursor proteins are biologically inactive in tests of coagulation. The precursor protein to prothrombin can be activated to thrombin nonphysiologically by snake venoms, demonstrating that the portion of the molecule necessary for this activity is intact. However, the prothrombin and other precursor proteins cannot bind divalent cations such as calcium and thus cannot interact with phospholipid-containing membranes, which are their normal sites of activation. This was puzzling for some time because abnormal prothrombin has the same number to amino acid residues and gives the same amino acid analysis after acid hydrolysis as does normal prothrombin. NMR studies revealed that normal prothrombin contains g-carboxyglutamate (see figure below), a previously unknown residue. The vitamin K-sensitive step in the synthesis of clotting factors is the carboxylation of ten or more glutamic acid residues at the amino-terminal end of the precursor protein to form g-carboxyglutamate. These amino acid residues are much stronger chelators of calcium than glutamate. The binding of Ca2+ by prothrombin anchors it to phospholipid membranes derived from blood platelets following injury. Th functional significance of the binding of prothrombin to phospholipid surfaces is that it brings prothrombin into close proximity with two proteins that mediate its conversion into thrombin-Factor Xa and Factor V. The amino-terminal fragment of prothrombin, which contains the Ca2+-binding sites, is released in this activation step. Thrombin free in this way from the phospholid surface can then activate fibrinogen in the plasma.
In hepatic microsomes, the reduction of vitamin K to its hydroquinone form (vitamin KH2) precedes the bicarbonate-dependent carboxylation of precursor prothrombin, descarboxyprothrombin, to prothrombin (see figure below). This carboxylase activity for the synthesis of prothrombin is linked to an epoxidase activity for vitamin KH2, which oxidizes the vitamin to vitamin K epoxide (KO). An epoxide reductase, which requires NADH, converts vitamin K epoxide back to vitamin KH2. This reaction is the site of action of the coumarins and the site of genetic resistance to the coumarins, which is also characterized by an increase requirement for vitamin K. Thus it is the recycling of the vitamin to its reduced active cofactor that results ultimately in decreased thrombin levels. The vitamin K analog chloro-K1 (in which the 2-methyl group of vitamin K1 is replaced by a chloro group) directly inhibits the carboxylase and epoxidase reaction, which are not sensitive to coumarins.
Vitamin K is reduced to the hydroquinone form (KH2). Stepwise oxidation to vitamin K epoxide (KO) is coupled to protein carboxylation, wherein descarboxyprothrombin is converted to prothrombin by carboxylation of glutamate residues to g-carboxyglutamate. Enzymatic reduction of the epoxide with NADH as a cofactor regenerates vitamin KH2. The oxidation of vitamin KH2. The oxidation of vitamin K is inhibited by the chloro analog of vitamin K epoxide is the coumarin-sensitive step.
Dicoumerol, the prototype of coumarin drugs, is of relatively low potency, with a slow onset of up to 5 days for peak activity and hypoprothrombinemia. The anticoagulant effect may persist for more than 1 week after stopping the drug. Even though over doses can be antidoted with vitamin K1, clinical adjustment of anticoagulation, particularly downward, is difficult. Warfarin has become the most widely used of the coumarin drugs. It is the most potent, with many patient being maintained on only 5 mg/day
Warfarin was initially introduced as a rodenticide because it was thought too dangerous for human use. It is still used in pest control. The drug, as sweetened pellets, causes rats to die from internal bleeding. In the early 1960s rats resistant to warfarin were noted in London. They were nicknamed "super rats." Several years later this phenomenon appeared in humans. It has been shown to be an inherited autosomal dominant trait. Person with this trait require a 20-fold increase in the drug to achieve anticoagulation-easily fatal to normal patients. Explanations for this unusual phenomenon have been proposed. One is that a tissue protein regulating the synthesis of one or more of the clotting factors became genetically altered. Another is a mutation in the enzyme daphorase that makes it less susceptible to coumarin drug inhibition.
Questions
1. What factors affect cardiac O2 supply and demand?
2. What are the symptoms and etiology of angina pectoris?
3. Both the nitrates and beta-adrenergic blocking agents have antianginal properties.
Compare and contrast the pharmacologic effects of these
two classes of compounds.
4. What are the sites of action of the nitrates?
5. When prescribing nitrates what would you advise a patient concerning adverse effects?
6. What advantages do the calcium-channel blockers have in the treatment of angina? What
is their mechanism of action? What are
contraindications to their use?
7. What are the two major objectives of drug therapy in angina pectoris?
8. What is the mechanism of action of nitroglycerine?
Other Topics
Hypercholesterolemia Drugs
Angiotensin-converting enzyme inhibitors
Antistimulants - histamine