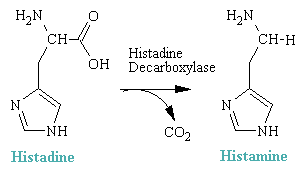
Histamine is 2-(4-imidazolyl)ethylamine. It is a hydrophilic molecule comprised of an imadazole ring and an amino group connected by two methylene groups. It arises in vivo by decarboxylation of the amino acid histadine.
IX. Histamines Antagonists and Local Anesthetics
Outline
A. Histamine
B. Histamine Antagonists
1. H1 Antagonists
2. H2 Antagonists
3. Proton Pump Inhibitors
4. Cromolyn Sodium
C. Local Anesthetics
A. Histamine
Histamine is 2-(4-imidazolyl)ethylamine. It is a hydrophilic molecule comprised of an imadazole ring and an amino group connected by two methylene groups. It arises in vivo by decarboxylation of the amino acid histadine.
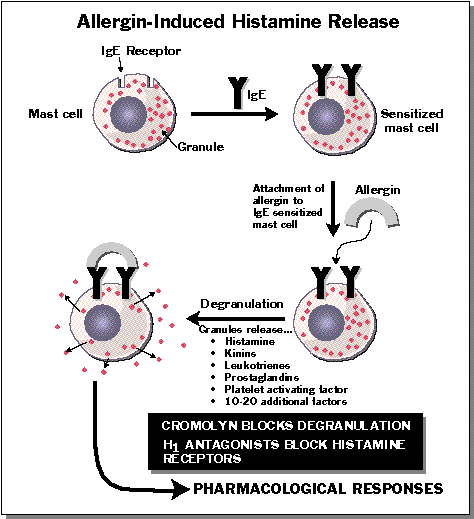
Histamine is concentrated in mast cells, cells whose function is essentially to release histamine and immunoglobins when tissue damage occurs. They are especially numerous in parts of the body that are injured often, such as the fingers and toes, or which enjoy frequent contact with the environment, such as the mucosa of the lips, nose, etc. Histamine is also a neurotransmitter in the CNS and a typical problem with some antihistamines is drowsiness. The effort has been to produce compounds that do not enter the brain very well.
There are many drugs with histamine-like properties, and most contain the following fragment:
:
Histamine contracts many smooth muscles, such as those of the bronchi and gut, but powerfully relaxes others, including those of fine blood vessels. It is also a very potent stimulus to gastric acid production. Effects attributable to these actions dominate the overall response to the drug; however, there are several others, of which edema formation and stimulation of sensory nerve endings are perhaps the most familiar. Some of these effects as bronchoconstriction and contraction of the gut, are mediated by one type of histamine receptor, the H1 receptors, which are readily blocked by pyrilamine and other such classical antihistamines, now more properly described as histamine H1-receptor blocking drugs of simply H1 blockers. Other effects, most notably gastric secretion, are completely refractory to such antagonists, involve activation of H2 receptors, and are susceptible to inhibition by the more recently developed histamine H2-receptor blocking drugs. Still others, such as the hypotension resulting from vascular dilation, are mediated by receptors of both H1 and H2 types, since they are annulled only by a combination of H1 and H2 blockers.
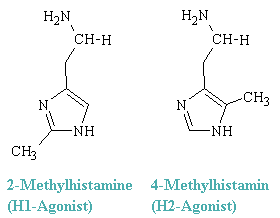
The two classes of histamine receptors also reveal themselves by differential responses to various histamine-like agonists. Thus, 2-methylhistamine preferentially elicits responses mediated by H1 receptors, whereas 4-methylhistamine has corresponingly preferential effect mediated through H2 receptors. These conpounds are representatives of two classes of histtamin-like dtugs, the H1-agonists and H2-agonists.
B. Histamine Antagonists
H1 Antagonists
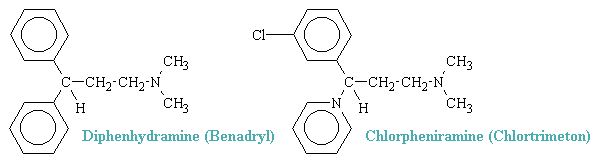
All of the available antagonists are reversable, competative inhibitors of the actions of histamine. The structure of almost all of the "classic" antihistamines have a tertiary amino group linked by two- or three-atom chain to two aromatic substituens and confrom to the general formula shown below, where Ar is aryl and X is a nitrogen or a carbon aton or a C-O- ether linkage.
Two common examples of H1 antagonists are shown below.
H1-blocking drugs have an established and valued place in the symptomatic treatment of various immediate hypersensitivity reactions, in which their usefulness is attributable to their antagonism of endogenously released histamine, one of several autoacids that elicit allergic response. In addition, the central properties of some of the series are of considerable therapeutic value in suppressing motion sickness.
In treating diseases of allergy, the effect of antihistamines is purely palliative and confined to the suppression in varying degree of symptoms attributable to the pharmacological activity of histamine released by the antigen-antibody reaction. The drugs do not diminish the intensity of this reaction, which is the root cause of the various hypersensitivity diseases. This limitation must be clearly recognized. In bronchial asthma, histamine blockers are singularly ineffectual. The have no role in the therapy of severe attacks in which chief reliance must be placed on epinephrine, isoproterenol, and theophylline. Equally, in the treat5ment of systemic anaphylaxis, in which autoacids other than histamine are again important, the mainstay of therapy is once more epinephrine, with histamine antagonists having only a subordinate and adjuvant role.
Other allergies of the respiratory tract are more amenable to therapy with H1 blockers. The best results are obtained in seasonal rhinitis (hay fever) and conjunctivitis, in which these drugs relieve the sneezing, rhinorrhea, and itching of the eyes, nose and throat.
H2 Antagonists
The H2 blockers are reversible, competitive antagonists of the actions of histamine on H2 receptors. They are highly selective in their action and are highly selective in their action and are virtually without effect on H1 receptors. The most prominent of the effects of histamine that are mediated by H2 receptors is stimulation of gastric acid secretion, and it is the ability of the H2 blockers to inhibit this effect that explains much of their importance. Despite the widespread distribution of H2 receptors in the body, H2 blockers interfere remarkably little with physiological function other than gastric secretion, implying that extragastric H2 receptors are of minor physiological importance
H2 blockers are used in treatment of peptic ulcer disease PUD; a disease in which ulceration occurs in the lower esophagus, stomach, duodenum, or jejunum. The most prominent symptom is gnawing pain that is relieved by food and alkali, but worsened by alcohol and condiments. The proximate cause of PUD is gastric acid hypersecretion.
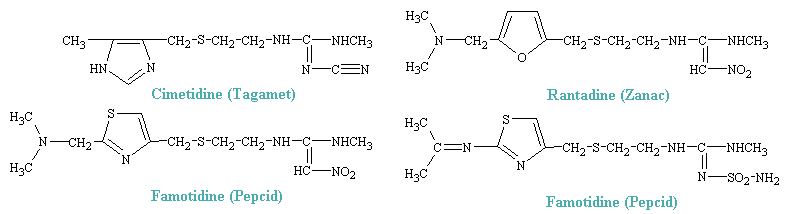
The synthesis of H2 antagonists was acheived by stepwise modifications of the histamine molecule, which resulted, some 200 compounds later, in the first highly effective drug with potent H2-blocking activity, burimamide. This, like later compounds, retained the imidazole ring of histamine byt possessed a much bulkier side chain. Cimetidine, the first H2 blocker to be introduced for general clinical use, won rapid acceptance for the treatment of ulcers and other gastric hypersecretory conditions and soon became one of the most widely prescribed of all drugs. this success led to the synthesis of numerous congeners. Some of the more popular drugs are shown below.
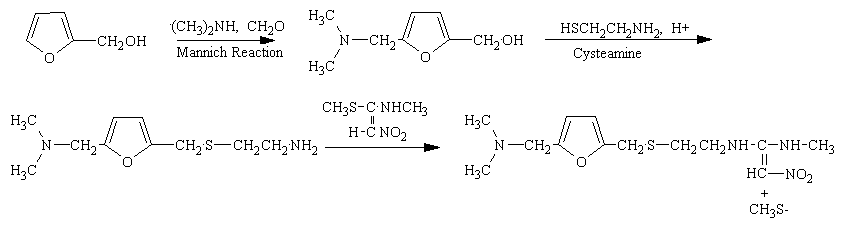
More Info On H2 Blockers 3-D Structure of Rantadine The synthesis of rantadine is not difficult. The figure below shows the three steps of its synthesis from common starting chemicals.
Proton Pump Inhibitors
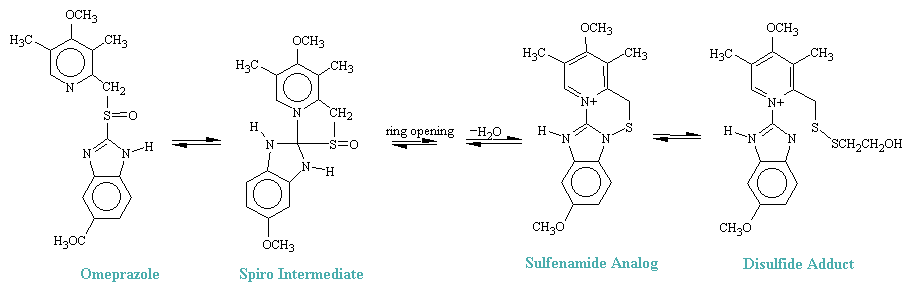
The parietal cells secrete acid by means of a membrane pump, identified as an H+, K+-ATPase, that exchanges hydrogen ions for potassium ions. By analogy with the familiar Na+, K+-ATPase, whose function can be inhibited by digitalis, this proton pump can likewise by inhibited by a newly discovered family. Omeprazole is the prototypical "acid pump" inhibitor which was allowed for clinical use in 1989. Its effects on gastric acid reduction are profound, showing a greater decrease of daily acid secretion than is obtained with four cimetidine given four times a day.
It has been demonstrated in vitro that under acid conditions as high as .5M, HCl, omeprazole cyclized reversibly to a spiro-dyhydroimidazole intermediate (see figure below), which opens to sulfenic acid (not isolated). Cyclization, by the loss of H2O leads to a cyclic sulfenamide that was isolated and identified. Treatment of the sulfenamide with mercaptoehanol (HSCH2CH2OH) opened the ring to produce the predicted disylfide adduct shown. Since these conditions simulate the gastric environment and H+, K+ATPase was known to have an essential -SH group, it has been proposed that the sulfenamide produced from omeprazole is the chemical species that forms a covalent drug-enzyme complex with H+, K-ATPase in the acid compartment of the parietal cell, thereby blocking H+ release.
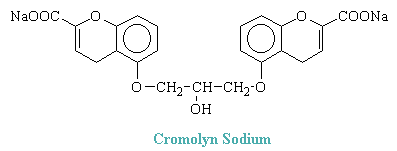
Cromolyn Sodium
Cromolyn sodium, the disodium salt of 1,3-bis(2-carboxychromone-5-yloxy)-2-hydroxypropane, has the following structure:
Cromolyn does not relax bronchial or other smooth muscle. Nor does it inhibit significantly responses to these muscles to any of a variety of pharmacological spasmogens. It does, however, inhibit the release of histamine and other autocoids (including leukotrienes) from human lung during allergic responses mediated by IgE antibodies and thereby the stimulus for bronchospasm. Inhibition of the liberation of leukotrienes is particularly important in allergic bronchial asthma, where these products appear to be the principal cause of bronchoconstriction. Cromolyn acts on the pulmonary mast cells for the immediate hypersensitivity reaction. Cromolyn does not inhibit the binding of IgE to mast cells nor the interaction between cell-bound IgE and specific antigen; rather, it suppresses the secretory response to this reaction.
Cromolyn sodium is insufflated into the lings by a special device as a micronized powder. The drug is strictly prophylactic; it will not abort an asthmatic attack in progress.
C. Local Anesthetics
The first local anesthetic to be discovered was cocaine, an alkaloid contained in large amounts in the leaves of Erythroxylon coca, a shrub growing in the Andes Mountains. Over 9 million kilograms of these leaves are consumed annually by the 2 million inhabitants of the highlands of Peru, who chew or suck the leaves for the sense of wellbeing it produces.
Local anesthetics are drugs that block nerve conduction when applied locally to nerve conduction when applied locally to nerve tissue in appropriate concentrations. They act on any part of the nervous system and on every type of nerve fiber. For example, when they are applied to the motor cortex impulse transmission from that area stops, and when they are injected into the skin they prevent the initiation and the transmission of sensory impulses. A local anesthetic in contact with a nerve trunk can cause both sensory and motor paralysis in the area innervated. The great practical advantage of the local anesthetic is that their action is reversible: their use is followed by complete recovery in nerve function with no evidence of structural damage to nerve fibers of cells.
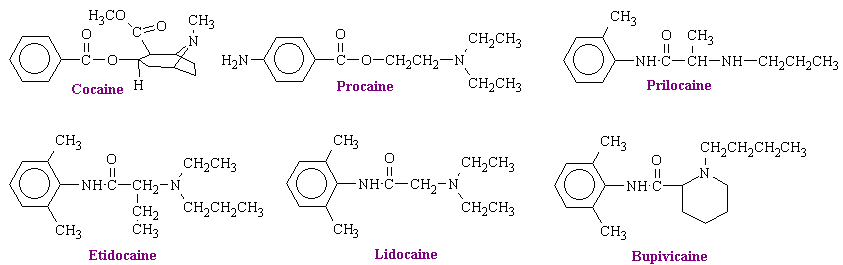
The structures some of the typical anesthetics are shown below.
These structures contain hydrophilic and hydrophobic domains that are separated by an intermediate alkyl chain. Linkage of these two domains is of either the ester or amide type. the ester link is important because this bond is readily hydrolyzed during metabolic degradation and inactivation in the body. Procaine, for example, can be divided into three main portions: the aromatic acid (para-aminobenzoic), the alcohol (ethanol), and the tertiary amino group (diethylamino). Changes in any part of the molecule alter the anesthetic potency and the toxicity of the compound. Increasing the length of the alcohol group leads to a greater anesthetic potency. It also leads to an increase in toxicity.
Local anesthetics prevent the generation and the conduction of the nerve impulse. Their site of action is the cell membrane. Local anesthetic and other classes of agents (e.g., alcohols and barbiturates) block conduction by decreasing or preventing the large transient increase in the permeability of the membrane to sodium ions that is produced by a slight depolarization of the membrane. As anesthetic action progressively develops in a nerve, the threshold for electrical excitability gradually increases and the safety factor for conduction decreases; when this action is sufficiently well developed, block of conduction is produced.
The local anesthetics also reduce the permeability of resting nerve to potassium as well as to sodium ions. Since changes in permeability to potassium require higher concentration of local anesthetic, blockade of conduction is not accompanied by any large or consistent change in the resting potential.
All the commonly used local anesthetics contain a tertiary or secondary nitrogen atom and, therefore, can exist either as the uncharged tertiary of secondary amine or as the positively charged substituted ammonium cation, depending on the dissociation content of the compound and the pH of the solutions. The pKa of a typical local anesthetic lies between 8.0 and 9.0, so that only 5 to 20% will be protonated at the pH of the tissues. this fraction, although small, is important because the drug usually has to diffuse through connective tissue and other cellular membranes to reach its site of action, and it is generally agreed that it can do so only in the form of the uncharged amine. Once the anesthetic has reached the nerve, the form of the molecule active in nerve fibers is the cation which combines with some receptor in the membreane to prevent the generationof an action potential.
Properties and Blockade Determinants Types and Descriptions of Ester Local Anesthetics Types amd Descriptions of Amide Anesthetics
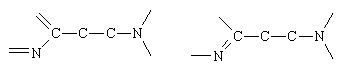
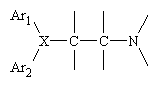
{kind=link}